Die Dissertation von Carsten Griebel beschäftigt sich mit der enzym-katalysierten kinetischen Racematspaltung von Tramadol-Analoga und deren industrieller Anwendbarkeit, erstellt an der RWTH Aachen zwischen 1998 und 2002. Besonderer Dank gilt verschiedenen Mitarbeitenden und Institutionen, die Unterstützung in Forschung und Analyse geleistet haben. Die Arbeit enthält umfassende theoretische und experimentelle Teile sowie zahlreiche Publikationen zu den durchgeführten Studien.








![VERZEICHNISSE IV
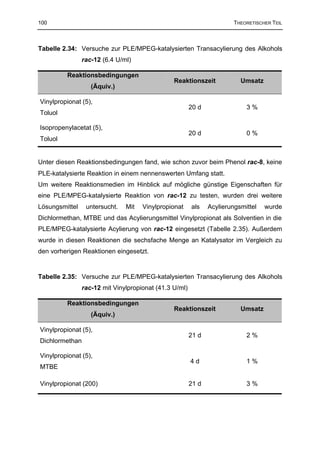
3.3.4.3 Darstellung von (±)-(1SR,2RS,4RS)-Essigsäure-3-dimethylaminomethyl-4-hydroxy-
4-(3-methoxy-phenyl)-cyclohexylester (rac-24) 201
3.3.4.4 Darstellung von (±)-(1SR,3RS,4RS)-Propionsäure-4-dimethylaminomethyl-3-hydroxy-
3-(3-methoxy-phenyl)-cyclohexylester (rac-45) 203
3.3.5 Versuche zur Darstellung von (±)-(1RS,5RS,8RS)-8-Dimethylaminomethyl-1-
(3-methoxy-phenyl)-2,4-dioxa-bicyclo-[3.3.1]-nonan-3-on (rac-48) 204
3.3.5.1 Versuch zur Darstellung von rac-48 durch Acylierung mit Bis-(trichlormethyl)-carbonat 204
3.3.5.2 Versuch zur Darstellung von rac-48 durch Acylierung mit 1,1´-Carbonyldiimidazol 205
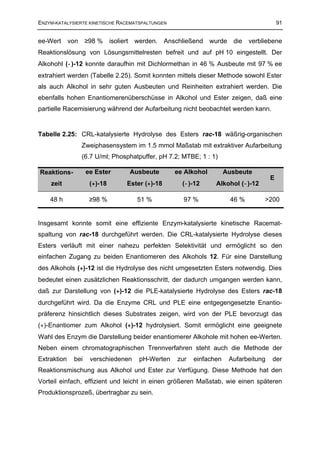
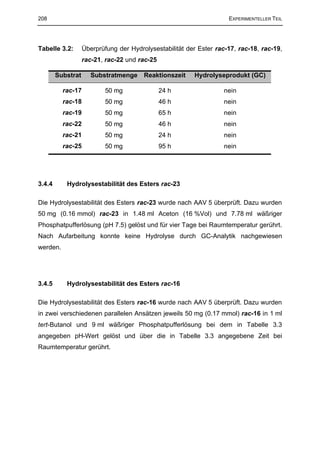
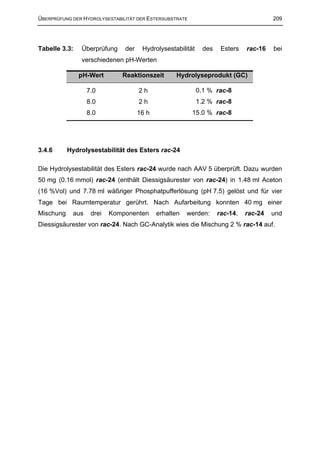
3.4 Überprüfung der Hydrolysestabilität der Estersubstrate 207
3.4.1 Allgemeine Arbeitsvorschrift zur Überprüfung der Hydrolysestabilität der racemischen
Substrat Ester (AAV 5) 207
3.4.2 Hydrolysestabilität des Esters rac-15 207
3.4.3 Hydrolysestabilität der Ester rac-17, rac-18, rac-19, rac-21, rac-22 und rac-25 207
3.4.4 Hydrolysestabilität des Esters rac-23 208
3.4.5 Hydrolysestabilität des Esters rac-16 208
3.4.6 Hydrolysestabilität des Esters rac-24 209
3.5 Bestimmung von Umsätzen und Enantiomerenverhältnissen 210
3.6 Enzym-katalysierte Racematspaltungen im wäßrigen und organischen
Medium 214
3.6.1 Allgemeine Arbeitsvorschriften 214
3.6.1.1 Allgemeine Arbeitsvorschrift zur enzymatischen Hydrolyse im wäßrigen Einphasen
System (AAV 6) 214
3.6.1.2 Allgemeine Arbeitsvorschrift zur enzymatischen Hydrolyse im wäßrig-organischen
Zweiphasen System (AAV 7) 214
3.6.1.3 Allgemeine Arbeitsvorschrift zur enzymatischen Transacylierung in organischen
Lösungsmitteln (AAV 8) 215
3.6.2 Berechnung des E-Werts 216
3.6.3 Enzymatische Hydrolysen von (±)-(1RS,2RS)-Buttersäure-3-(2-dimethylaminomethyl-
1-hydroxy-cyclohexyl)-phenylester (rac-15) 217
3.6.3.1 PLE-katalysierte Hydrolyse des Esters rac-15 im wäßrigen Einphasensystem unter
Zusatz von Aceton als Cosolvens 217
3.6.3.2 PLE-katalysierte Hydrolyse des Esters rac-15 im wäßrigen Einphasensystem 217
3.6.3.3 PLE-katalysierte Hydrolyse des Esters rac-15 im wäßrigen Einphasensystem zur
Darstellung von (+)-15 218
3.6.3.4 Versuch zur AChE-katalysierten Hydrolyse des Esters rac-15 im wäßrigen
Einphasensystem 219
3.6.3.5 CAL-katalysierte Hydrolyse des Esters rac-15 im wäßrigen Einphasensystem 219
3.6.3.6 Mucor miehei Lipase-katalysierte Hydrolyse des Esters rac-15 im wäßrigen
Einphasensystem 220
3.6.3.7 HLE-katalysierte Hydrolyse des Esters rac-15 im wäßrigen Einphasensystem 220
3.6.3.8 CE-katalysierte Hydrolyse des Esters rac-15 im wäßrigen Einphasensystem 221](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-9-320.jpg)




























![24 THEORETISCHER TEIL
Aufarbeitung analysiert werden konnten. Mittels dieser effektiven Analytik ließen sich
detaillierte Informationen über den gesamten Reaktionsverlauf einer enzymatischen
Reaktion unter Einsatz geringer Substanz- und vor allem Enzymmengen gewinnen.
2.2.1 Synthesewege der von GRÜNENTHAL bereitgestellten Substanzen96
Die von der GRÜNENTHAL GmbH verwendeten Synthesewege zur Darstellung der
cyclohexanoiden Ausgangsmaterialien rac-14⋅HCl97, rac-12⋅HCl98 und rac-13⋅HCl
basieren auf der in Kap. 1.2 beschriebenen Tramadol-Syntheseroute, so daß viele
Erfahrungen aus diesem Produktionsprozeß in die Reaktionen eingebracht werden
konnten.
O CH3 1) BrMg OMe
O
N Cl
H 3C N(Me)2
O THF
O O H 3C Cl O O 2) Trennung der
Diastereomeren
MeCN 3) H
OH OH
OMe NaBH4 OMe
HO
O Me2N Me2N
rac-27 rac-14
Abb. 2.3: Syntheseroute der GRÜNENTHAL GmbH zur Darstellung von rac-14.
Zur Einführung der neuen sekundären Hydroxylgruppe wurde anstelle von
Cyclohexanon ein als Monoketal geschütztes 1,4- bzw. 1,3-Diketon in die Mannich-
Reaktion eingesetzt (Abb. 2.3 und Abb. 2.4). Aus der Mannich-Reaktion des
3,3-Dimethyl-1,5-dioxa-spiro[5.5]undecan-8-on (26, Abb. 2.4) wurde nur das
gewünschte 9-Alkylierungsprodukt erhalten. Das unerwünschte 7-Alkylierungs-
produkt wurde wahrscheinlich aufgrund von sterischer Hinderung nicht gebildet. Die
erhaltenen jeweiligen Mannich-Basen wurden danach in einer Grignard-Reaktion mit
dem Grignard-Reagenz des 3-Bromanisols umgesetzt. Es wurden in beiden Fällen
cis /trans Mischungen erhalten, aus denen die gewünschten cis-Isomere abgetrennt
wurden und nach Abspaltung der Ketal-Schutzgruppen mit Mineralsäure die Ketone](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-38-320.jpg)




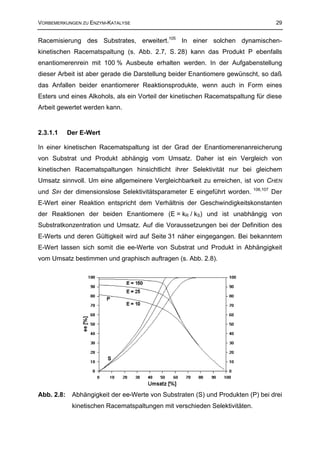
![30 THEORETISCHER TEIL
Aus Abb. 2.8 geht hervor, daß das Substrat (S) bei einer kinetischen Racemat-
spaltung prinzipiell immer bei hinreichend großen Umsätzen auf Kosten der
Ausbeute enantiomerenrein erhalten werden kann. Das Produkt (P) hingegen kann
nur bei sehr selektiven Reaktionen mit E-Werten größer als 100 nahezu
enantiomerenrein erhalten werden.
Zur Bestimmung von E müssen zwei der drei Variablen: Umsatz, ee-Wert des
Substrates und ee-Wert des Produkts bekannt sein und in eine der aufgeführten
Formeln eingesetzt werden6,107,108:
ln [1− c(1+ eeP )]
Formel 2.1: E= , für c < 50 %
ln [1 − c(1− eeP )]
ln [(1− c)(1− ee S )]
Formel 2.2: E= , für c > 50 %
ln [(1− c)(1+ ee S )]
1− ee S ee P (1− ee S )
ln ln (ee + ee )
1 + (eeS /ee P ) S
E= =
P
Formel 2.3:
1 + ee S ee P (1+ ee S )
ln ln
1 + (eeS /ee P ) ee P + ee S )
(mit: c = Umsatz, ee P = ee-Wert des Produkts, ee S = ee-Wert des Substrats)
KAZLAUSKAS et al. haben eine schnelle photospektroskopische Methode zur
Bestimmung des E-Werts entwickelt, die auf der Umsetzung der einzelnen
Enantiomeren basiert (“Quick E-Wert“).109 Häufig stehen gerade diese aber nicht zur
Verfügung.
E-Werte sind für sehr kleine und sehr große Umsätze mit einem großen Fehler
behaftet. Entweder läßt sich c nur mit beschränkter Genauigkeit bestimmen oder die
gefundenen ee-Werte sind sehr groß, so daß relativ kleine Fehler bei Bestimmungen
von c, eeS oder eeP einen großen Einfluß auf den berechneten E-Wert haben, da sie
logarithmisch in die Berechnung eingehen.6,21,108,110 Die Angabe von E-Werten
größer als 100 ist aus demselben Grund wenig sinnvoll, da hier ebenfalls der Einfluß
kleiner Meßfehler sehr groß ist.6,108,110,111 In einigen Fällen können sogar E-Werte um
die 50 nur mit einer Genauigkeit von ± 10 angegeben werden (Angabe als E ≈ 50).6
Häufig ist in der Praxis die Bestimmung von Enantiomerenüberschüssen genauer als
die Bestimmung des Umsatzes, bei der das Verhältnis zweier verschiedener Stoffe](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-44-320.jpg)

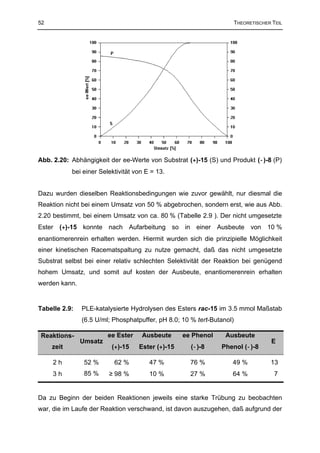
![32 THEORETISCHER TEIL
Hydrolysereaktionen führen.116 Für Gleichgewichtsreaktionen wurde von CHEN und
SIH ebenfalls eine Methode zur Bestimmung von E unter Kenntnis der
Gleichgewichtskonstanten K entwickelt (Formel 2.4).107 Wie aus Abb. 2.9 hervorgeht,
fallen bei reversiblen, kinetischen Racematspaltungen die ee-Werte des Substrats
bei Umsätzen über 50 % deutlich ab. Nicht umgesetztes Substrat kann also im Falle
einer reversiblen Reaktion nicht mehr enantiomerenrein erhalten werden. Hohe
ee-Werte des Substrats bei großen Umsätzen deuten hingegen auf eine extreme
Gleichgewichtslage (K ≈ 0, Fall a in Abb. 2.9) hin.
ln [1 − (1 + K)(c + ee S (1 − c))] ln [1 - (1 + K) c (1 + eeP )]
Formel 2.4: E= =
ln [1 − (1 + K)(c − ee S (1 − c))] ln [1 - (1 + K) c (1 - eeP )]
(mit: c = Umsatz, ee P = ee-Wert des Produkts, ee S = ee-Wert des Substrats)
Abb. 2.9: Abhängigkeit der ee-Werte a) des Produkts und b) des Substrats bei
reversiblen kinetischen Racematspaltungen mit E = 100 für verschiedene
Gleichgewichtskonstanten K (K = a: 0, b: 0.1, c: 0.5, d: 1, e: 5).
ANTHONSEN et al. haben einen einfachen Weg zur Bestimmung E und K entwickelt,
indem sie in einem regressiven mathematischen Verfahren über eine Serie von eeS-
und eeP-Werten eine Fit E-Wert Funktion berechnen.117
c) Substrat- oder Produktinhibierung
Bei dem Vorliegen von Produktinhibierung kann sich der E-Wert über den
Reaktionsverlauf ändern.118,119 Um den E-Wert zu beeinflussen, muß allerdings eine
enantioselektive Inhibierung vorliegen118, die sogar eine Rückreaktion des Produkts](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-46-320.jpg)
























![ENZYM-KATALYSIERTE KINETISCHE RACEMATSPALTUNGEN 59
60 % der nicht umgesetzte Ester (+)-15 enantiomerenrein vorliegt und in einer
maximalen Ausbeute von ca. 40 % erhalten werden kann.
Es konnte somit gezeigt werden, daß eine Enzym-katalysierte Hydrolyse eines
Esters auf Tramadol-Basis unter Einbezug einer phenolischer Hydroxylgruppe mit
hoher Selektivität und Aktivität möglich ist. Dies, obwohl die vom Enzym
anzugreifende phenolische Hydroxylgruppe mehrere C-Atome von den Stereo-
zentren der Verbindungen entfernt ist.
Die Bestimmung der Absolutkonfiguration der Enantiomeren des Phenols 8 erfolgte
durch Vergleich der Drehwerte mit bereits publizierten.91,183 Von der Grünenthal
GmbH ist für das Hydrochlorid des (+)-(R,R)-3-(2-Dimethylaminomethyl-1-hydroxy-
cyclohexyl)-phenols ein Drehwert von [α ] 20 = + 5.4º (c = 1.0, MeOH) veröffentlicht
D
worden.91 Hierbei ist die Bestimmung der Absolutkonfiguration des (+)-8⋅HCl durch
Röntgenstrukuranalyse erfolgt.184
Die Bestimmung der Absolutkonfigurationen der Ester 15 und 16 erfolgte in Analogie
zu der des Phenols 8.
2.4.1.2 Enzym-katalysierte Hydrolysen von (±)-(1RS,2RS)-Essigsäure-3-
(2-dimethylaminomethyl-1-hydroxy-cyclohexyl)-phenylester (rac-16)
Neben den Effekten des Cosolvens auf die Enzym-katalysierte Hydrolyse von Estern
des O-Desmethyltramadols (rac-8), ist auch der Einfluß der Acylgruppe des
Substrats auf die Aktivität oder Selektivität der enzymatischen Reaktion von
Interesse.
Im allgemeinen stellt für Lipasen die einzige Limitierung hinsichtlich des akzeptierten
Substratspektrums ihre begrenzte Eignung für die Umsetzung raumerfüllender
Acylgruppen dar: gemäß dem Vorbild der Triglyceride werden lineare aliphatische
Acylgruppen besser umgesetzt als z.B. Arene.101,110,185](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-73-320.jpg)























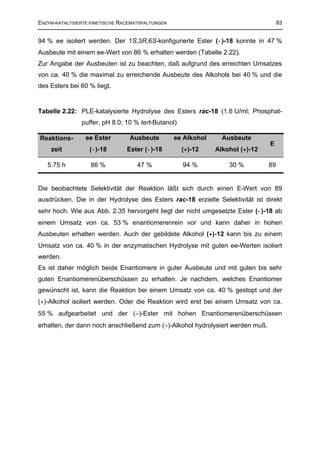
















![102 THEORETISCHER TEIL
geschwindigkeit bei einem Zusatz von 0.2 Vol% Wasser beobachtet worden.85 Daher
wurde für diesen Versuch mit dem Katalysator PLE/BSA/MPEG ein Wasserzusatz
von 0.25 % gewählt.
Tabelle 2.36: Versuche zur PLE/BSA/MPEG-katalysierten Transacylierung des
Alkohols rac-12
Reaktionsbedingungen
Reaktionszeit Umsatz
(Äquiv.)
PLE/BSA/MPEG (24.4 U/ml),
11 d 0%
Vinylpropionat (5), Dichlormethan
PLE/BSA/MPEG (20.0 U/ml), H2O (0.25 %),
11 d 0%
Vinylpropionat (5), Toluol
Im Laufe der Reaktionszeit änderte sich die Farbe des PLE/BSA-Katalysators von
weiß in ein dunkles rot-braun. Dies ist ein typisches Merkmal dafür, daß der Zusatz
BSA mit Carbonyl-Nebenprodukten reagiert. Diese Nebenprodukte können nur aus
der enzymatischen Hydrolyse des Vinylesters stammen. Eine derartige
Nebenreaktion in größerem Umfang ist ebenfalls von Ruppert84 wie auch anderen213
beobachtet worden.
O
+E
R1 OH
+ H2O A
O +E O
+ [R1-CO-E]
R1 O
B
+ R2-OH
O + H2O
O
R2 + E + R2-OH
R1 O R1 OH
C
Abb. 2.43: Hydrolyse des Acyldonors als Nebenreaktion der Transacylierung.](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-116-320.jpg)


































































![ALLGEMEINE VORBEMERKUNGEN 169
Polarimetrie
Gerät: Perkin-Elmer-Polarimeter PE 241. Die Messung der Drehwerte erfolgte bei
20 ºC in einer Mikroküvette (Länge 10 cm) mit der Natriumdoppellinie (λ = 589.0 und
589.6 nm). Die Angabe der spezifischen Drehung [α ] 20 erfolgte in (grd⋅dm3)/(dm⋅g),
D
die Angabe der Konzentration c in (g/dm3).
Autotitrator
Gerät: STAT-Titrino 718 der Firma METROHM. Das Gerät wurde im pH-STAT Modus
mit NaOH-Maßlösungen (1 N und 0.1 N) der Firma MERCK betrieben.
Ultrafiltration
Gerät: Ultrafiltrationszelle Amicon Modell 2800 mit einer Membran PM 30
(Ausschlußgrenze: >30 kDa) von MILLIPORE.
Gefriertrocknung
Gerät: Christ Alpha 2-4.
Schmelzpunkte
Gerät: Büchi-Schmelzpunktapparatur SMP-20.
Die Schmelzpunkte wurden unkorrigiert angegeben.
Siedepunkte
Die Siedepunkte wurden unkorrigiert angegeben.
Dünnschichtchromatographie
Es wurden DC-Alufolien Kieselgel 60 F254 der Firma MERCK mit einer Schichtdicke
von 0.25 mm verwendet. Die Detektion erfolgte durch UV-Fluoreszenzlöschung bei
λ = 254 nm sowie durch Anfärben mit einer Lösung aus Eisessig / dest. H2O / konz.
H2SO4 / p-Anisaldehyd (in einem Volumenverhältnis von 75 : 2 : 1.5 : 1) und an-
schließendem Erhitzen.
Präparative Säulenchromatographie
Es wurde Kieselgel 60, 0.062 - 0.100 mm, der Firma MERCK verwendet.](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-183-320.jpg)








![178 EXPERIMENTELLER TEIL
3.3.2 Darstellung der racemischen Substrate zur enzymatischen
Transacylierung
3.3.2.1 Darstellung von (±)-(1RS,2RS)-2-Dimethylaminomethyl-1-(3-methoxy-
phenyl)-cyclohexanol (rac-7)
Die Base rac-7 wurde nach AAV 1 aus ihrem Hydrochlorid freigesetzt, dazu wurden
10 g (33.3 mmol) rac-7⋅HCl in 16 ml dest. Wasser und 12 ml Dichlormethan
suspendiert und mit 4.4 ml 40 %iger NaOH-Lösung versetzt. Nach Aufarbeitung
wurden 8.70 g (33.0 mmol, 99 %) rac-7 als farbloser Sirup erhalten.
OH
10
5 6 1 9 11
O
3 7 15
4 2 14
N 12
13
8
rac-7: MW = 263.4 g⋅mol-1
H-NMR (300 MHz, CDCl3): δ = 7.2 ppm (dd, 1 H, 3J = 8 Hz, 3J = 8 Hz, 1 H, 13-H),
1
7.2 (s, 1 H, 10-H), 7.0 (d, 3J = 8 Hz, 1 H, 12-H), 6.8 (dm, 3J = 8 Hz, 1 H, 14-H), 3.8 (s,
3 H, 15-H), 2.4 (dd, 3J=4 Hz, 2J = 13 Hz, 1 H, 7´-H), 2.1 - 2.0 (m, 7 H, 8-H, 7-H), 1.9 -
1.5 (m, 8 H, 3-H, 4-H, 5-H, 6-H), 1.4 - 1.2 (m, 1 H, 2-H).
13
C-NMR (75.4 MHz, CDCl3): δ = 159.5 ppm [u] (C-11), 152.0 [u] (C-9), 128.9 [d]
(C-13), 117.4 [d] (C-14), 111.2 [d] (C-12), 111.0 [d] (C-10), 76.9 [u] (C-1), 61.6 [u]
(C-7), 55.2 [d] (C-15), 47.7 [d] (C-8), 44.8 [d] (C-2), 41.3 [u] (C-6), 27.9 [u] (C-3), 26.9
[u] (C-4), 22.3 [u] (C-5).
13
Die Zuordnung der C-NMR-Signale war durch einen Vergleich mit Literaturdaten
eindeutig möglich.245](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-192-320.jpg)
![DARSTELLUNG DER SUBSTRATE ZUR ENZYMATISCHEN RACEMATSPALTUNG 179
3.3.2.2 Darstellung von (±)-(1RS,2RS)-3-(2-Dimethylaminomethyl-1-hydroxy-
cyclohexyl)-phenol (rac-8)91
Zu 5.2 g (19.8 mmol) rac-7, gelöst in 9 ml abs. Toluol, wurden bei Raumtemperatur
40 ml 20 %ige Diisobutylaluminiumhydrid-Lösung (39.6 mmol) in abs. Toluol
zugetropft, wobei es zu einer leicht exothermen Reaktion und einer Gasentwicklung
kam. Anschließend wurde für 15 Stunden unter Rückfluß erhitzt. Nach Abkühlung auf
Raumtemperatur wurden unter Eiskühlung 11.3 ml Ethanol so zugetropft, daß eine
Innentemperatur von 15 ºC nicht überschritten wurde. Danach wurde 15 Min.
nachgerührt. Ebenfalls unter Eiskühlung wurden daraufhin 11.3 ml eines
Ethanol/Wasser-Gemisches (1 : 1) wieder so zugetropft, daß eine Innentemperatur
von 15 ºC nicht überschritten wurde. Anschließend wurde die Reaktionslösung mit
ca. 20 ml Toluol verdünnt und danach noch eine Stunde bei Raumtemperatur
nachgerührt. Der ausgefallene Feststoff wurde abfiltriert und mit fünf Volumenteilen
Essigsäureethylester bei 60°ºC für 30 Min. nachextrahiert. Nach Filtration wurden die
vereinigten organischen Phasen über Natriumsulfat getrocknet und am
Rotationsverdampfer unter vermindertem Druck eingeengt. Es wurden 4.592 g
(18.4 mmol, 93 %) eines farblosen Öls erhalten, welches farblose Kristalle mit einem
Schmelzpunkt von 139 ºC bildete.
OH
10
5 6 1 9 11
OH
3 7
4 2 14
N 12
13
8
rac-8: MW = 249.4 g⋅mol-1
H-NMR (300 MHz, CDCl3): δ = 7.5 ppm (s, 1 H, 10-H), 7.2 (dd, 3J = 8 Hz, 3J = 8 Hz,
1
1 H, 13-H), 6.8 (m, 2 H, 12-H, 14-H), 2.4 (dd, 3J = 4 Hz, 2J = 14 Hz, 1 H, 7´-H), 2.2 -
1.4 (m, 15 H, 3-H, 4-H, 5-H, 6-H, 7-H, 8-H), 1.4 - 1.2 (m, 1 H, 2-H).
13
C-NMR (75.4 MHz, CDCl3): δ = 157.0 ppm [u] (C-11), 151.0 [u] (C-9), 129.4 [d]
(C-13), 116.0 [d] (C-14), 113.3 [d] (C-12), 113.0 [d] (C-10), 78.3 [u] (C-1), 61.7 [u]
(C-7), 47.6 [d] (C-8), 44.4 [u] (C-2), 41.0 [u] (C-6), 27.9 [u] (C-3), 26.7 [u] (C-4), 22.2
[u] (C-5).](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-193-320.jpg)
![180 EXPERIMENTELLER TEIL
MS (EI, 70 eV) m/z (%): 249 (M+, 18), 121 (3), 58 (100), 45 (6).
MS (CI, Isobutan, 100 eV) m/z (%):306 ([M + C4H9]+, 4), 250 ([M + H]+, 100), 249 (M+,
10).
IR (KBr): ∼ = 3201 (s), 2985 (s), 2863 (s), 2834 (s), 2789 (s), 2425 (w), 1280 (s),
ν
1252 (m), 1215 (s), 1166 (m), 1118 (m), 1098 (m), 1079 (m), 1033 (w), 10006 (w),
988 (s), 963 (m), 905 (w), 865 (m), 846 (m), 763 (m), 707 (m) cm-1.
C15H23NO2 (249.2 g⋅mol-1): berechnet gefunden
C 72.25 71.98
H 9.30 9.10
N 5.62 5.56
3.3.2.3 Darstellung von (±)-(1RS,2RS)-3-(3-Dimethylamino-1-hydroxy-1,2-
dimethyl-propyl)-phenol (rac-11)
Die Base rac-11 wurde nach AAV 1 aus ihrem Hydrochlorid freigesetzt, dazu wurden
5.0 g (19.3 mmol) rac-11⋅HCl in 19 ml dest. Wasser und 2 ml Dichlormethan
suspendiert und mit 10 ml 40 %iger NaOH-Lösung versetzt. Nach Aufarbeitung
wurden 4.188 g (18.8 mmol, 97 %) rac-11 als farbloser Sirup erhalten, welcher
farblose, weiße Kristalle mit einem Schmelzpunkt von 98 ºC bildete.
OH
8
1 7 9
4 OH
3 5
2 12
N 10
11
6
rac-11: MW = 223.3 g⋅mol-1
H-NMR (300 MHz, CDCl3): δ = 8.3 – 8.1 (br s, 1 H, 9-OH), 7.21 (s, 1 H, 8-H), 7.06 (t,
1
3
J = 8 Hz, 1 H, 11-H), 6.82 (d, 3J = 8 Hz, 1 H, 10-H/12-H), 6.61 (dd, 3J = 8 Hz, 4J =
2 Hz, 1 H, 10-H/12-H), 2.67 (dd, 3J = 11 Hz, 2J = 13 Hz, 1 H, 5-H´), 2.31 (s, 6 H, 6-H),](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-194-320.jpg)
![DARSTELLUNG DER SUBSTRATE ZUR ENZYMATISCHEN RACEMATSPALTUNG 181
2.23 (dd, 3J = 3 Hz, 2J = 13 Hz, 1 H, 5-H), 2.06 (m, 1 H, 2-H), 1.49 (s, 3 H, 4-H), 0.65
(d, 3J = 7 Hz, 3 H, 3-H).
C-NMR (75.4 MHz, CDCl3): δ = 156.5 ppm [u] (C-9), 149.4 [u] (C-7), 128.5 [d]
13
(C-11), 117.2 [d] (C-12), 113.9 [d] (C-10), 113.7 [d] (C-8), 78.4 [u] (C-1), 64.1 [u]
(C-5), 45.6 [d] (C-6), 40.1 [d] (C-2), 21.8 [d] (C-4), 14.8 [d] (C-3).
MS (EI, 70 eV) m/z (%): 223 (M+, 10), 181 (3), 138 (3), 121 (11), 107 (6), 95 (4), 93
(10), 91 (5), 86 (69), 77 (9), 71 (18), 65 (14), 58 (100), 45 (62).
IR (KBr): ∼ = 3225 (s), 2981 (s), 2955 (s), 2884 (s), 2838 (s), 2802 (s), 2653 (s),
ν
2567 (m), 1615 (m), 1585 (s), 1507 (s), 1485 (s), 1467 (s), 1282 (s), 1255 (s), 1240
(s), 1260 (s), 1176 (m), 1162 (w), 1123 (s), 1103 (w), 1077 (w), 876 (s), 841 (s), 793
(s), 743 (m), 707 (s), 688 (w), 545 (w), 531 (w), 487 (w) cm-1.
C13H21NO2 (223.3 g⋅mol-1): berechnet gefunden
C 69.92 69.73
H 9.48 9.56
N 6.27 6.23
3.3.2.4 Darstellung von (±)-(1RS,3SR,6RS)-6-Dimethylaminomethyl-1-
(3-methoxy-phenyl)-cyclohexan-1,3-diol (rac-12)
Die Base rac-12 wurde nach AAV 1 aus ihrem Hydrochlorid freigesetzt, dazu wurden
3.293 g (10.4 mmol) rac-12⋅HCl in 5 ml dest. Wasser und 20 ml Dichlormethan
suspendiert und mit ca. 3.0 ml 40 %iger NaOH-Lösung versetzt. Nach Aufarbeitung
wurden 2.883 g (10.3 mmol, 99 %) rac-12 als farbloser, zäher Sirup erhalten, der
unter gelindem Erwärmen in einen Achatmörser überführt wurde und dort
anschließend erstarrte. 2.273 g (8.1 mmol, 79 %) wurden so als farblose Kristalle mit
einem Schmelzpunkt von 49 ºC gewonnen und 0.552 g (2.0 mmol, 19 %) als
farbloser Sirup zurückbehalten.](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-195-320.jpg)
![182 EXPERIMENTELLER TEIL
OH
5 10
6 1 9 11
HO O
3 7 15
4 2 14
N 12
13
8
rac-12: MW = 279.4 g⋅mol-1
H-NMR (400 MHz, CDCl3): δ = 7.24 ppm (t, 1 H, 13-H), 7.10 (s, 1 H, 10-H), 7.01 (d,
1
3
J = 7 Hz, 1 H, 12-H), 6.75 (ddd, 3J = 9 Hz, 4J = 3 Hz, 4J = 1 Hz, 1 H, 14-H), 4.12 (m,
1 H, 5-H), 3.80 (s, 3 H, 15-H), 2.50 – 2.75 (br s, 1 H, 1-OH), 2.38 (dd, 2J = 14 Hz,
3
J = 4 Hz, 1 H, 7-H´), 2.25 – 1.20 (m, 14 H, 2-H, 3-H, 4-H, 6-H, 7-H, 8-H).
13
C-NMR (100 MHz, CDCl3): δ = 159.6 [u] (C-11), 150.7 [u] (C-9), 129.1 [d] (C-13),
117.2 [d] (C-14), 111.6 [d] (C-12), 110.8 [d] (C-10), 78.6 [u] (C-1), 67.5 [d] (C-5), 60.7
[u] (C-7), 55.2 [d] (C-15), 50.0 [u] (C-6), 47.8 [d] (C-8), 44.3 [d] (C-2), 35.7 [u] (C-4),
26.3 [u] (C-3).
MS (EI, 70 eV) m/z (%): 279 (M+, 64), 234 (5), 135 (11), 107 (4), 84 (3) 58 (100).
IR (in CHCl3): ∼ = 3371 (s), 3079 (s), 2945 (s), 2860 (s), 2831 (s), 2784 (s), 1600 (s),
ν
1583 (s), 1484 (s), 1463 (s), 1432 (s), 1365 (m), 1315 (m), 1287 (s), 1255 (s), 1206
(m), 1158 (s), 1096 (s), 1074 (s), 1048 (s), 1012 (m), 979 (m), 959 (m), 861 (m), 844
(m), 829 (w), 782 (s), 756 (s), 730 (m), 704 (s), 666 (m), 569 (w) cm-1.
C16H25NO3 (279.4 g⋅mol-1): berechnet gefunden
C 68.79 68.44
H 9.02 9.38
N 5.01 4.93](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-196-320.jpg)
![DARSTELLUNG DER SUBSTRATE ZUR ENZYMATISCHEN RACEMATSPALTUNG 183
3.3.2.5 Darstellung von (±)-(1RS,3RS,6RS)-6-Dimethylaminomethyl-1-
(3-methoxy-phenyl)-cyclohexan-1,3-diol (rac-13)
Die Base rac-13 wurde nach AAV 1 aus ihrem Hydrochlorid freigesetzt, dazu wurden
2.422 g (7.7 mmol) rac-13⋅HCl in 10 ml dest. Wasser und 30 ml Dichlormethan
suspendiert und mit ca. 1.5 ml 40 %iger NaOH-Lösung versetzt. Nach Aufarbeitung
wurden 2.092 g (7.5 mmol, 98 %) rac-13 als weißer Feststoff mit einem
Schmelzpunkt von 106 ºC erhalten.
OH OH
10
5 6 1 9 11
O
3 7 15
4 2 14
N 12
13
8
rac-13: MW = 279.4 g⋅mol-1
H-NMR (400 MHz, CDCl3): δ = 7.99 ppm (s, 1 H, 1-OH), 7.26 (t, 3J = 8 Hz, 1 H,
1
13-H), 7.09 (s, 1 H, 10-H), 6.99 (s, 1 H, 14-H), 6.75 (m, 1 H, 12-H), 5.15 (s, 1 H,
5-OH), 4.06 (s, 1 H, 5-H), 3.82 (s, 3 H, 15-H), 2.47 (m, 2 H, 7-H, 3-H), 2.15 (d, 1 H,
7-H´), 2.12 (s, 6 H, 8-H), 2.05 (m, 1 H, 4-H), 1.97 (ddd, 4J = 2.75 Hz, 3J = 2.75 Hz,
2
J = 14.6 Hz, 1 H, 6-H), 1.92 (m, 1 H, 2-H), 1.75 (dd, 1 H, 2J = 14.6 Hz, 3J = 3.05 Hz,
6-H´), 1.60 – 1.65 (m, 2 H, 4-H´, 3-H´).
13
C-NMR (100 MHz, CDCl3): δ = 159.6 ppm [u] (C-11), 150.2 [u] (C-9), 129.1 [d]
(C-13), 117.0 [d] (C-14), 111.5 [d] (C-12), 110.8 [d] (C-10), 79.0 [u] (C-1), 67.0 [d]
(C-5), 61.3 [u] (C-7), 55.1 [d] (C-15), 47.8 [d] (C-8), 44.9 [u] (C-6), 44.0 [d] (C-2), 33.6
[u] (C-4), 21.8 [u] (C-3).
MS (EI, 70 eV) m/z (%): 279 (M+, 17), 135 (5), 58 (100), 45 (4).
IR (KBr): ∼ = 3375 (s), 3046 (m), 2969 (s), 2942 (s), 2874 (s), 2826 (s), 1598 (s),
ν
1488 (s), 1461 (s), 1438 (s), 1406 (w), 1377 (w), 1326 (m), 1285 (s), 1263 (s), 1246
(s), 1206 (m), 1177 (m), 1158 (s), 1113 (m), 1083 (w), 1044 (s), 1011 (m), 986 (m),
971 (m), 956 (m), 926 (w), 883 (s), 852 (s), 835 (m) 789 (s), 706 (s), 597 (m) cm-1.](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-197-320.jpg)
![184 EXPERIMENTELLER TEIL
C16H25NO3 (279.4 g⋅mol-1): berechnet gefunden
C 68.79 68.65
H 9.02 9.25
N 5.01 4.93
3.3.2.6 Darstellung von (±)-(1RS,2RS,4SR)-2-Dimethylaminomethyl-1-
(3-methoxy-phenyl)-cyclohexan-1,4-diol (rac-14)
Die Base rac-14 wurde nach AAV 1 aus ihrem Hydrochlorid freigesetzt, dazu wurden
1.802 g (5.7 mmol) rac-14⋅HCl in 10 ml dest. Wasser und 30 ml Dichlormethan
suspendiert und mit ca. 0.9 ml 40 %iger NaOH-Lösung versetzt. Nach Aufarbeitung
wurden 1.578 g (5.5 mmol, 99 %) rac-14 als farbloser, zäher Sirup erhalten, der
unter gelindem Erwärmen in einen Achatmörser überführt wurde und dort
anschließend erstarrte. 1.300 g (4.7 mmol, 82 %) wurden so als farblose Kristalle mit
einem Schmelzpunkt von 44 ºC gewonnen und 0.278 g (1 mmol, 17 %) als farbloses
sirupöses Öl zurückbehalten.
OH
10
5 6 1 9 11
O
HO 3 7 15
4 2 14
N 12
13
8
rac-14: MW = 279.4 g⋅mol-1
H-NMR (400 MHz, CDCl3): δ = 7.25 ppm (t, 3J = 8 Hz, 1 H, 13-H), 7.13 (s, 1 H,
1
10-H), 7.01 (s, 1 H, 12-H), 6.78 (ddd, 3J = 8 Hz, 4J = 3 Hz, 4J = 1 Hz, 1 H, 14-H), 3.81
(s, 3 H, 15-H), 3.75 (m, 1 H, 4-H), 2.7 - 2.9 (br s, OH), 2.42 (dd, 2J = 14 Hz,
3
J = 4.5 Hz, 1 H, 7-H´), 2.22 – 1.60 (m, 14 H, 2-H, 3-H, 5-H, 6-H, 7-H, 8-H).
13
C-NMR (100 MHz, CDCl3): δ = 159.7 ppm [u] (C-11), 151.0 [u] (C-9), 129.3 [d]
(C-13), 117.5 [d] (C-14), 111.7 [d] (C-12), 111.3 [d] (C-10), 76.2 [u] (C-1), 71.2 [d]
(C-4), 61.5 [u] (C-7), 55.5 [d] (C-15), 48.1 [d] (C-8), 43.5 [d] (C-2), 39.7 [u] (C-6), 37.2
[u] (C-3), 31.9 [u] (C-5).](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-198-320.jpg)

![186 EXPERIMENTELLER TEIL
7´-H), 2.1 - 1.4 (m, 17 H, 3-H, 4-H, 5-H, 6-H, 7-H, 8-H, 17-H), 1.4 - 1.2 (m, 1 H, 2-H),
1.0 (t, 3J = 5 Hz, 3 H, 18-H).
C-NMR (75.4 MHz, CDCl3): δ = 172.1 ppm [u] (C-15), 152.2 [u] (C-11), 150.8 [u]
13
(C-9), 128.8 [d] (C-13), 122.2 [d] (C-14), 119.0 [d] (C-12), 118.6 [d] (C-10), 76.9 [u]
(C-1), 61.5 [u] (C-7), 47.7 [d] (C-8), 44.7 [d] (C-2), 41.3 [u] (C-6), 36.2 [u] (C-16), 27.9
[u] (C-3), 26.8 [u] (C-4), 22.2 [u] (C-5), 18.4 [u] (C-17), 13.7 [d] (C-18).
MS (EI, 70 eV) m/z (%): 319 (M+, 9), 58 (100).
IR (KBr): ∼ = 3400 (w), 3091 (m), 2975 (s) 2945, 2856 (s), 2829 (s), 1755 (s), 1608
ν
(m), 1588 (m), 1461 (m), 1445 (m), 1434 (s), 1417 (s), 1383 (m), 1156 (s), 1156 (s),
1096 (s), 808 (m), 708 (m) cm-1.
C19H29NO3 (319.4 g⋅mol-1): berechnet gefunden
C 71.44 71.03
H 9.15 9.19
N 4.38 4.35
3.3.3.2 Darstellung von (±)-(1RS,2RS)-Essigsäure-3-(2-dimethylaminomethyl-
1-hydroxy-cyclohexyl)-phenylester (rac-16)
Die Synthese erfolgte gemäß AAV 2, dazu wurden 2.490 g (10.0 mmol) rac-8 mit
einer Mischung aus 1.318 g (12.9 mmol) Essigsäureanhydrid und 0.08 g (1.0 mmol)
Pyridin in 40 ml Dichlormethan umgesetzt. Nach Aufarbeitung und
Säulenchromatographie (EE : MeOH = 3 : 1 über Kieselgel, Rf(rac-16)= 0.26;
Rf (rac-8) = 0.10) wurden 2.881 g (9.9 mmol, 99 %) eines weißen kristallinen
Feststoffs mit einem Schmelzpunkt von 84 ºC erhalten.](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-200-320.jpg)
![DARSTELLUNG DER SUBSTRATE ZUR ENZYMATISCHEN RACEMATSPALTUNG 187
OH
10
5 6 1 9 11
O 15 16
7
4 3 2 14
N 12
O
13
8
rac-16: MW = 291.4 g⋅mol-1
H-NMR (500 MHz, CDCl3): δ = 7.3 ppm (m, 3 H, 13-H, 10-H, 12-H/14-H), 6.93 (m,
1
1 H, 12-H/14-H), 5.8 – 6.4 (br s, 1 H, 1-OH), 2.38 (dd, 3J = 4 Hz, 2J = 14 Hz, 1 H,
7´-H), 2.29 (s, 3 H, 16-H), 2.25 – 1.25 (m, 16 H, 2-H, 3-H, 4-H, 5-H, 6-H, 7-H, 8-H).
13
C-NMR (75.4 MHz, CDCl3): δ = 169.3 ppm [u] (C-15), 152.0 [u] (C-11), 150.5 [u]
(C-9), 128.7 [d] (C-13), 122.2 [d] (C-14), 118.9 [d] (C-12), 118.4 [d] (C-10), 77.3 [u]
(C-1), 61.5 [u] (C-7), 47.7 [d] (C-8), 44.7 [d] (C-2), 41.3 [u] (C-6), 27.9 [u] (C-3), 26.8
[u] (C-4), 22.2 [u] (C-5), 21.1 [d] (C-16).
MS (EI, 70 eV) m/z (%): 291 (M+, 31), 121 (3), 58 (100) 45 (8).
IR (KBr): ∼ = 3076 (w), 3007 (w), 2981 (w), 2947 (s), 2924 (s), 2856 (m), 2828 (m),
ν
2786 (m), 1755 (s), 1609 (w), 1588 (w), 1482 (w), 1459 (m), 1436 (m), 1377 (m),
1293 (w), 1268 (w), 1213 (s), 1154 (m), 1140 (w), 1094 (w), 1085 (w), 1022 (m), 991
(m), 965 (m), 938 (w), 889 (w), 839 (w), 806 (w), 784 (w), 710 (w) cm-1.
C17H25NO3 (291.4 g⋅mol-1): berechnet gefunden
C 70.07 69.91
H 8.65 8.13
N 4.81 4.71
3.3.3.3 Darstellung von (±)-(1RS,2RS)-Buttersäure-3-(3-dimethylamino-1-
hydroxy-1,2-dimethyl-propyl)-phenylester (rac-17)
Die Synthese erfolgte gemäß AAV 2, dazu wurden 1.786 g (8.0 mmol) rac-11 mit
einer Mischung aus 1.52 g (9.6 mmol) Buttersäureanhydrid und 0.06 g (0.8 mmol)
Pyridin in 20 ml Dichlormethan umgesetzt. Nach Aufarbeitung und](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-201-320.jpg)
![188 EXPERIMENTELLER TEIL
Säulenchromatographie (EE : MeOH = 2 : 1, 1 % Triethylamin über Kieselgel,
Rf (rac-17)= 0.58) wurden 1.995 g (6.8 mmol, 85 %) eines farblosen Öls erhalten.
OH 8
1 7 9 14 16
4 O 13
5
3 2 12 15
N 10 O
11
6
rac-17: MW = 293.4 g⋅mol-1
H-NMR (400 MHz, CDCl3): δ = 7.24 (s, 1 H, Ar), 7.22 (s, 1 H, Ar), 7.15 (s, 1 H, Ar),
1
6.87 (m, 1 H, 11-H), 2.57 (dd, 3J = 11 Hz, 2J = 13 Hz, 1 H, 5-H´), 2.44 (t, 3J = 7 Hz,
2 H, 14-H), 2.22 (s, 6 H, 6-H), 2.14 (dd, 3J = 3 Hz, 2J = 13 Hz, 1 H, 5-H), 1.94 (m, 1 H,
2-H), 1.69 (m, 2 H, 15-H), 1.40 (s, 3 H, 4-H), 0.96 (t, 3J = 7 Hz, 3 H, 16-H), 0.70 (d, 3J
= 7 Hz, 3 H, 3-H).
13
C-NMR (100 MHz, CDCl3): δ = 170.8 ppm [u] (C-13), 149.5 [u] (C-9), 149.3 [u] (C-
7), 127.3 [d] (C-11), 122.0 [d] (C-12), 118.3 [d] (C-10), 118.1 [d] (C-8), 76.2 [u] (C-1),
63.0 [u] (C-5), 44.7 [d] (C-6), 39.3 [d] (C-2), 35.2 [u] (C-14), 20.9 [d] (C-4), 17.4 [u]
(C-15), 14.8 [d] (C-3), 12.6 [d] (C-16).
MS (EI, 70 eV) m/z (%): 293 (M+, 4), 86 (7), 84 (12), 71 (4), 58 (100), 45 (7).
IR (kapillar): ∼ = 2971 (s), 2877 (m), 2827 (m), 2784 (m), 1759 (s), 1608 (m), 1586
ν
(m), 1466 (s), 1370 (m), 1236 (m), 1153 (s), 1098 (w), 1080 (w), 1029 (m), 947 (m),
838 (w), 704 (m) cm-1.
C17H27NO3 (293.4 g⋅mol-1): berechnet gefunden
C 69.59 69.52
H 9.28 8.93
N 4.77 4.67](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-202-320.jpg)
![DARSTELLUNG DER SUBSTRATE ZUR ENZYMATISCHEN RACEMATSPALTUNG 189
3.3.3.4 Darstellung von (±)-(1SR,3RS,4RS)-Buttersäure-4-dimethylamino-
methyl-3-hydroxy-3-(3-methoxy-phenyl)-cyclohexylester (rac-18)
Die Synthese erfolgte gemäß AAV 3 bei Raumtemperatur, dazu wurden 3.265 g
(10.3 mmol) rac-12⋅HCl, gelöst in 33 ml abs. THF, mit 2.950 g (26.3 mmol) Kalium-
tert-butylat über einen Zeitraum von 10 Min. mit 1.66 ml (1.703 g, 16.0 mmol)
Buttersäurechlorid, gelöst in 3 ml abs. THF, versetzt. Nach Aufarbeitung und
Säulenchromatographie (Diisopropylether : MeOH = 1:1 über Kieselgel,
Rf (rac-18) = 0.41; Rf (rac-12) = 0.18) wurden 2.915 g (8.3 mmol, 81 %) eines klaren
sirupösen Öls erhalten.
O OH
18 10
5 6 1 9 11
19 16 O O
17 3 7 15
4 2 14
N 12
13
8
rac-18: MW = 349.5 g⋅mol-1
H-NMR (400 MHz, CDCl3): δ = 7.26 ppm (t, 3J = 8 Hz, 1 H, 13-H), 7.14 (s, 1 H,
1
10-H), 7.03 (s, 1 H, 12-H), 6.75 (dd, 3J = 8 Hz, 4J = 3 Hz, 1 H, 14-H), 5.21 (m, 1 H,
5-H), 3.82 (s, 3 H, 15-H), 2.42 (dd, 2J = 14 Hz, 3J = 4 Hz, 1 H, 7-H´), 2.30 – 1.40 (m,
18 H, 2-H, 3-H, 4-H, 6-H, 7-H, 8-H, 17-H, 18-H), 0.91 (t, 3J = 7 Hz, 3 H, 19-H).
13
C-NMR (100 MHz, CDCl3): δ = 173.8 ppm [u] (C-16), 159.8 [u] (C-11), 150.6 [u]
(C-9), 129.3 [d] (C-13), 117.4 [d] (C-14), 111.9 [d] (C-12), 111.0 [d] (C-10), 78.4 [u]
(C-1), 71.1 [d] (C-5), 60.5 [u] (C-7), 55.1 [d] (C-15), 47.7 [d] (C-8), 46.0 [u] (C-6), 44.1
[d] (C-2), 36.4 [u] (C-17), 32.2 [u] (C-4), 25.9 [u] (C-3), 18.5 [u] (C-18), 13.6 [d]
(C-19).
MS (EI, 70 eV) m/z (%): 350 ([M+H]+, 9), 349 (M+, 38), 262 (7), 135 (8), 58 (100), 45
(3).
IR (kapillar): ∼ = 2947 (s), 2864 (m), 2831 (s), 2784 (m), 1730 (s), 1600 (m), 1583
ν
(m), 1484 (s), 1462 (s), 1432 (m), 1383 (m), 1358 (m), 1306 (m), 1288 (s), 1256 (s),](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-203-320.jpg)

![DARSTELLUNG DER SUBSTRATE ZUR ENZYMATISCHEN RACEMATSPALTUNG 191
C-NMR (100 MHz, CDCl3): δ = 172.7 ppm [u] (C-16), 159.7 [u] (C-11), 149.0 [u]
13
(C-9), 129.3 [d] (C-13), 117.7 [d] (C-14), 112.0 [d] (C-12), 111.6 [d] (C-10), 76.1 [u]
(C-1), 70.5 [d] (C-5), 61.1 [u] (C-7), 55.5 [d] (C-15), 46.6 [d] (C-8), 43.4 [u] (C-6), 43.0
[d] (C-2), 37.0 [u] (C-17), 29.6 [u] (C-4), 22.4 [u] (C-3), 18.9 [u] (C-18), 14.0 [d]
(C-19).
MS (EI, 70 eV) m/z (%): 349 (M+, 16), 135 (6), 58 (100).
IR (kapillar): ∼ = 3584 (m), 3079 (s), 2944 (m), 2873 (m), 2766 (m), 1733 (s), 1601
ν
(m), 1584 (m), 1485 (m), 1461 (s), 1432 (m), 1381 (w), 1287 (s) 1255 (s), 1201 (s),
1188 (s), 1166 (s), 1104 (m), 1088 (m), 1049 (m), 985 (w), 960 (w) 860 (w), 825 (w),
782 (w), 734 (w), 702 (w) cm-1.
C20H31NO4 (349.5 g⋅mol-1): berechnet gefunden
C 68.74 68.35
H 8.94 9.08
N 4.01 4.30
3.3.3.6 Darstellung von (±)-(1RS,3RS,6RS)-3-(6-Dimethylaminomethyl-1,3-
dihydroxy-cyclohexyl)-phenol (rac-20)91
Zu 1.790 g (6.4 mmol) rac-13, gelöst in 8 ml abs. Toluol, wurden bei
Raumtemperatur 42 ml Diisobutylaluminiumhydrid-Lösung (1 M, 42.0 mmol) in abs.
Toluol zugetropft, wobei es zu einer leicht exothermen Reaktion und einer starken
Gasentwicklung kam. Anschließend wurde die Reaktionslösung für 1 Stunde bei
Raumtemperatur und danach für weitere 24 Stunden unter Rückfluß erhitzt. Nach
Abkühlung auf Raumtemperatur wurden unter Eiskühlung 5.0 ml Ethanol so
zugetropft, daß eine Innentemperatur von 15 ºC nicht überschritten wurde. Danach
wurde 15 Min. nachgerührt. Ebenfalls unter Eiskühlung wurden anschließend 10.0 ml
eines Ethanol/Wasser-Gemisches (1 : 1) wieder so zugetropft, daß eine
Innentemperatur von 15 ºC nicht überschritten wurde. Nach Filtration wurde die](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-205-320.jpg)
![192 EXPERIMENTELLER TEIL
organische Phase über Natriumsulfat getrocknet und am Rotationsverdampfer unter
vermindertem Druck eingeengt. Es wurden 1.653 g (5.6 mmol, 88 %) eines farblosen
Feststoffs mit einem Schmelzpunkt von 74 ºC erhalten, welcher noch Spuren rac-13
enthielt. Eine chromatographische Abtrennung dieser Verunreinigung war nicht
möglich, daher wurde im folgenden die in Spuren verunreinigte Substanz eingesetzt.
OH OH
10
5 6 1 9 11
OH
3 7
4 2 14
N 12
13
8
rac-20: MW = 265.4 g⋅mol-1
H-NMR (400 MHz, CDCl3): δ = 7.18 ppm (mt, 3J = 8 Hz, 1 H, 13-H), 7.1 – 6.8 (br s,
1
1 H, 11-OH), 6.98 (s, 1 H, 10-H), 6.80 (s, 1 H, 14-H), 6.64 (dd, 1 H, 3J = 8 Hz,
4
J = 2 Hz, 12-H), 6.0 – 6.1 (br s, 2 H, 1-OH, 5-OH), 4.05 (m, 1 H, 5-H), 2.50 – 1.45
(m, 15 H, 2-H, 3-H, 4-H, 6-H, 7-H, 8-H).
13
C-NMR (100 MHz, CDCl3): δ = 157.0 ppm [u] (C-11), 150.0 [u] (C-9), 129.6 [d]
(C-13), 116.3 [d] (C-14), 113.9 [d] (C-10), 112.5 [d] (C-12), 79.4 [u] (C-1), 67.8 [d]
(C-5), 61.6 [u] (C-7), 48.1 [d] (C-8), 45.1 [u] (C-6), 44.1 [d] (C-2), 33.6 [u] (C-4), 22.3
[u] (C-3).
MS (EI, 70 eV) m/z (%): 349 (M+, 16), 135 (6), 58 (100).
IR (kapillar): ∼ = 3584 (m), 3079 (s), 2944 (m), 2873 (m), 2766 (m), 1733 (s), 1601
ν
(m), 1584 (m), 1485 (m), 1461 (s), 1432 (m), 1381 (w), 1287 (s) 1255 (s), 1201 (s),
1188 (s), 1166 (s), 1104 (m), 1088 (m), 1049 (m), 985 (w), 960 (w) 860 (w), 825 (w),
782 (w), 734 (w), 702 (w) cm-1.](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-206-320.jpg)
![DARSTELLUNG DER SUBSTRATE ZUR ENZYMATISCHEN RACEMATSPALTUNG 193
3.3.3.7 Darstellung von (±)-(1RS,3RS,6RS)-Buttersäure-3-(6-dimethylamino-
methyl-1,3-dihydroxy-cyclohexyl)-phenylester (rac-21)95
1.190 g (4.5 mmol) rac-20 wurde zu einer Lösung aus 50 ml dest. Wasser und 6.42 g
NaHCO3 (76.4 mmol) bei Raumtemperatur gegeben. Nach einer Stunde hatte sich
eine klare Lösung gebildet, zu der innerhalb von 15 Minuten tropfenweise 6.65 ml
(6.40 g, 40.5 mmol) Buttersäureanhydrid unter starker Gasentwicklung gegeben
wurden. Anschließend wurde noch 15 Min. bei Raumtemperatur zur
Vervollständigung der Reaktion gerührt und danach dreimal mit jeweils 15 ml
Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden über NaSO4
getrocknet. Nach Einengen am Rotationsverdampfer wurden 1.446 g braunes,
sirupöses Rohprodukt erhalten. Nach Säulenchromatographie (EE : MeOH = 2 : 1
über Kieselgel, Rf = 0.33; Rf (rac-13) = 0.26) konnten 1.417 g (4.2 mmol, 93 %)
rac-21 als schwach hellbrauner Feststoff mit einem Schmelzpunkt von 79 ºC erhalten
werden.
OH OH
10
5 6 1 9 11 16 18
O 15
3 7 17
4 2 14
N 12
O
13
8
rac-21: MW = 335.4 g⋅mol-1
H-NMR (400 MHz, CDCl3): δ = 7.27 ppm (t, 3J = 8 Hz, 1 H, 13-H), 7.20 ( br s, 1 H, -
1
H), 7.09 (m, 1 H, -H), 6.88 (dd, 3J = 9 Hz, 4J = 2 Hz, 1 H, -H), 4.01 (mt, 3J = 3 Hz,
1 H, 5-H), 2.5 – 1.5 (m, 19 H, 2-H, 3-H, 4-H, 6-H, 7-H, 8-H, 16-H, 17-H), 0.97 (t,
3
J = 7 Hz, 3 H, 18-H).
13
C-NMR (100 MHz, CDCl3): δ = 172.2 ppm [u] (C-15), 151.0 [u] (C-11), 150.3 [u]
(C-9), 129.3 [d] (C-13), 122.2 [d] (C-14), 119.7 [d] (C-12), 118.7 [d] (C-10), 78.7 [u]
(C-1), 67.2 [d] (C-5), 61.4 [u] (C-7), 47.5 [d] (C-8), 46.1 [u] (C-6), 44.0 [d] (C-2), 36.5
[u] (C-16), 33.6 [u] (C-4), 22.1 [u] (C-3), 18.8 [u] (C-17), 14.0 [d] (C-18).
MS (EI, 70 eV) m/z (%): 265 (M+, 14), 121 (3), 58 (100) 45 (6).](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-207-320.jpg)
![194 EXPERIMENTELLER TEIL
IR (KBr): ∼ = 3371 (m), 3169 (s9, 2946 (s), 2830 (s), 2786 (m), 1615 (m) 1585 (m),
ν
1481 (s), 1424 (s), 1369 (w), 1352 (m), 1305 (m), 1293 (s), 1278 (s) 1251 (s), 1221
(m), 1165 (m), 1108 (s), 1075 (s), 1033 (m), 1007 (m), 984 (s), 952 (s), 904 (m), 874
(m), 863 (m), 841 (s), 782 (s), 734 (m), 703 (s), 579 (w), 469 (w) cm-1.
3.3.3.8 Darstellung von (±)-(1SR,2RS,4RS)-Buttersäure-3-dimethylamino-
methyl-4-hydroxy-4-(3-methoxy-phenyl)-cyclohexylester (rac-22)
Die Synthese erfolgte gemäß AAV 3 bei Raumtemperatur, dazu wurden 2.761 g
(8.7 mmol) rac-14⋅HCl, gelöst in 28 ml abs. THF, mit 1.500 g (13.4 mmol) Kalium-
tert-butylat und über einen Zeitraum von 10 Min. mit 1.410 ml (1.447 g, 13.6 mmol)
Buttersäurechlorid, gelöst in 2 ml abs. THF, versetzt. Nach Aufarbeitung und
Säulenchromatographie (Diisopropylether : MeOH = 1 : 1 über Kieselgel, Rf = 0.39;
Rf (rac-14) = 0.14) wurden 2.396 g (6.9 mmol, 79 %) eines klaren sirupösen Öls
erhalten.
OH
10
5 6 1 9 11
17 O
19 16 O 3 7 15
4 2 14
18
N 12
13
O 8
rac-22: MW = 349.5 g⋅mol-1
H-NMR (400 MHz, CDCl3): δ = 7.18 ppm (t, 3J = 8 Hz, 1 H, 13-H), 7.05 (s, 1 H,
1
10-H), 6.93 (s, 1 H, 12-H), 6.68 (ddd, 3J = 8 Hz, 4J = 3 Hz, 4J = 1 Hz, 1 H, 14-H), 4.78
(m, 1 H, 4-H), 3.74 (s, 3 H, 15-H), 2.35 (dd, 2J = 14 Hz, 3J = 4 Hz, 1 H, 7-H´), 2.25 –
1.50 (m, 18 H, 2-H, 3-H, 5-H, 6-H, 7-H, 8-H, 17-H, 18-H), 0.89 (t, 3J = 7 Hz, 3 H,
19-H).
13
C-NMR (100 MHz, CDCl3): δ = 173.5 ppm [u] (C-16), 159.8 [u] (C-11), 150.8 [u]
(C-9), 129.3 [d] (C-13), 117.4 [d] (C-14), 111.9 [d] (C-12), 111.1 [d] (C-10), 76.1 [u]
(C-1), 73.3 [d] (C-4), 61.3 [u] (C-7), 55.5 [d] (C-15), 48.1 [d] (C-8), 43.4 [d] (C-2), 39.4
[u] (C-6), 36.9 [u] (C-17), 33.3 [u] (C-3), 27.9 [u] (C-5), 18.9 [u] (C-18), 14.0 [d]
(C-19).](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-208-320.jpg)

![196 EXPERIMENTELLER TEIL
H-NMR (400 MHz, CDCl3): δ = 7.19 ppm (t, 3J = 8 Hz, 1 H, 13-H), 7.05 (s, 1 H,
1
10-H), 6.93 (s, 1 H, 14-H), 6.70 (dd, 3J = 8 Hz, 4J = 2.5 Hz, 1 H, 12-H), 6.08 (br s,
1 H, 1-OH), 4.78 (m, 1 H, 4-H), 3.74 (s, 3 H, 15-H), 2.4 – 1.5 (m, 19 H, 2-H, 3-H, 5-H,
6-H, 7-H, 8-H, 17-H, 18-H), 1.25 (m, 4 H, 19-H, 20-H), 0.83 (t, 3J = 7 Hz, 3 H, 21-H).
13
C-NMR (100 MHz, CDCl3): δ = 173.7 ppm [u] (C-16), 159.8 [u] (C-11), 150.6 [u]
(C-9), 129.4 [d] (C-13), 117.4 [d] (C-14), 111.9 [d] (C-12), 111.2 [d] (C-10), 76.0 [u]
(C-1), 73.2 [d] (C-4), 61.3 [u] (C-7), 55.5 [d] (C-15), 47.9 [d] (C-8), 43.2 [d] (C-2), 39.4
[u] (C-6), 34.9 [u] (C-17) 33.3 [u] (C-3), 31.6 [u] (C-19), 27.9 [u] (C-5), 25.1 [u] (C-18),
22.7 [u] (C-20), 14.3 [d] (C-21).
MS (EI, 70 eV) m/z (%): 377 (M+, 10), 135 (3), 58 (100).
MS (CI, Isobutan, 100 eV) m/z (%): 434 ([M + C4H9]+, 3), 378 ([M + H]+, 100), 262 (4).
IR (kapillar): ∼ = 2955 (s), 2861 (s), 2832 (m), 2783 (m), 1729 (s), 1601 (m),1583
ν
(m), 1484 (s), 1464 (s), 1432 (s), 1380 (m), 1355 (m), 1317 (m), 1288 (s), 1251 (s),
1176 (s), 1137 (m), 1097 (m), 1049 (m), 1034 (s), 1014 (s), 783 (m), 736 (w), 703 (m)
cm-1.
C22H35NO4 (377.5 g⋅mol-1): berechnet gefunden
C 69.99 70.15
H 9.34 9.49
N 3.71 3.61](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-210-320.jpg)
![DARSTELLUNG DER SUBSTRATE ZUR ENZYMATISCHEN RACEMATSPALTUNG 197
3.3.3.10 Darstellung von (±)-(1SR,2RS,4RS)-Essigsäure-3-dimethylamino-
methyl-4-hydroxy-4-(3-methoxy-phenyl)-cyclohexylester (rac-24)
Die Synthese erfolgte gemäß AAV 3 bei 0 ºC, dazu wurden 1.700 g (5.4 mmol)
rac-14⋅HCl, gelöst in 20 ml Diethylether auf 0 ºC gekühlt, mit 1.510 g (13.45 mmol)
Kalium-tert-butylat und mit 0.630 g (8.0 mmol) Essigsäurechlorid, gelöst in 2.6 ml
Diethylether, versetzt. Nach Aufarbeitung wurden 1.816 g eines gelben, öligen
Rohprodukts erhalten. Nach Säulenchromatographie (Diisopropylether : MeOH =
1 : 1 über Kieselgel, Rf = 0.46; Rf (rac-14) = 0.19) wurden 1.292 g einer gelblich
öligen Mischung aus rac-24 und eines Diesters (Rf = 0.51) aus rac-14 und
Essigsäure (Verhältnis nach den Signalen der Protonen der Methoxygruppe (15-H)
im 1H-NMR: 4 : 1) erhalten. Ein Versuch zur weiteren Aufreinigung durch präparative
HPLC (Acetonitril : Wasser = 3 : 1 über RP-Select B-LiChrosor b-Säule) führte zur
Zersetzung von rac-24. Eine weitere Aufreinigung von rac-24 war somit nicht
möglich. Im folgenden wurde daher die nach Säulenchromatographie erhaltene
Mischung eingesetzt.
OH
10
5 6 1 9 11
O
17 16 O 3 7 15
4 2 14
N 12
13
O 8
rac-24: MW = 263.4 g⋅mol-1
H-NMR (400 MHz, CDCl3): δ = 7.18 ppm (t, 3J = 8 Hz, 1 H, 13-H), 7.10 (s, 1 H,
1
10-H), 6.90 (s, 1 H, 12-H), 6.70 (md, 3J = 8 Hz, 1 H, 14-H), 4.77 (m, 1 H, 4-H), 3.75
(s, 3 H, 15-H), 2.34 (md, 2J = 14 Hz, 1 H, 7-H´), 2.30 – 1.45 (m, 17 H, 2-H, 3-H, 5-H,
6-H, 7-H, 8-H, 17-H).
13
C-NMR (100 MHz, CDCl3): δ = 170.9 ppm [u] (C-16), 159.8 [u] (C-11), 150.8 [u]
(C-9), 129.3 [d] (C-13), 117.4 [d] (C-14), 111.8 [d] (C-12), 111.1 [d] (C-10), 76.0 [u]
(C-1), 73.7 [d] (C-4), 61.3 [u] (C-7), 55.5 [d] (C-15), 48.2 [d] (C-8), 43.3 [d] (C-2), 39.4
[u] (C-6), 33.3 [u] (C-3), 27.9 [u] (C-5), 21.8 [d] (C-17).](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-211-320.jpg)
![198 EXPERIMENTELLER TEIL
3.3.3.11 Darstellung von (±)-(1RS,3RS,4RS)-Buttersäure-3-butyryloxy-4-
dimethylaminomethyl-3-(3-methoxy-phenyl)-cyclohexylester (rac-25)
Zu einer Mischung von 1.750 g (6.3 mmol) rac-13 und 0.250 g (0.5 mmol) Pyridin in
5 ml Dichlormethan wurden unter Rühren 1.090 g (6.9 mmol) Buttersäureanhydrid
gegeben. In einer schwach exothermen Reaktion verfärbte sich die Reaktionslösung
hierbei schwach gelb. Die Reaktionslösung wurde für 26 Stunden bei
Raumtemperatur gerührt, bis durch DC-Kontrolle kein Edukt mehr detektiert werden
konnte. Anschließend wurde die schwach gelbe Reaktionslösung mit einem
Überschuß ges. NaHCO3-Lösung für weitere 20 Stunden kräftig gerührt. Nach
Phasentrennung wurde dreimal mit jeweils 15 ml Dichlormethan extrahiert. Die
vereinigten organischen Phasen wurden mit dest. Wasser gewaschen und über
NaSO4 getrocknet. Nach Einengen am Rotationsverdampfer wurde ein gelbliches,
Sirup ähnliches Öl erhalten, das im Hochvakuum von Pyridinresten befreit wurde.
Nach Säulenchromatographie (Diisopropylether : MeOH = 1 : 1 über Kieselgel,
Rf = 0.48) wurden 1.916 g (4.6 mmol, 67 %) rac-25 erhalten.
O O
18 22
21
16 O O 20 23
19 17
1 10
5 6 9 11
O
3 7 15
4 2 14
N 12
13
8
rac-25: MW = 419.6 g⋅mol-1
H-NMR (400 MHz, CDCl3): δ = 7.24 ppm (t, 3J = 8 Hz, 1 H, 13-H), 6.76 (m, 3 H,
1
10-H, 12-H, 14-H), 5.18 (m, 1 H, 5-H), 3.80 (s, 3 H), 15-H), 3.09 (dd, 2J = 15 Hz,
4
J = 3 Hz, 1 H, 6-H´), 2.46 – 2.30 (m, 4 H, 6-H, 7-H´,17-H/21-H), 2.22 (t, 3J = 7 Hz,
2 H, 17-H/21-H), 2.09 (s, 6 H, 8-H), 1.95 – 1.60 (m, H, 2-H, 3-H, 4-H, 7-H, 18-H,
22-H), 1.01 (t , 3J = 7 Hz, 3 H, 19-H/23-H), 0.95 (t , 3J = 7 Hz, 3 H, 19-H/23-H).
13
C-NMR (100 MHz, CDCl3): δ = 172.6 ppm [u] (C-16), 171.9 [u] (C-20), 159.1 [u]
(C-11), 143.0 [u] (C-9), 128.8 [d] (C-13), 117.9 [d] (C-14), 112.4 [d] (C-12), 111.4 [d]
(C-10), 84.0 [u] (C-1), 69.0 [d] (C-5), 59.0 [u] (C-7), 55.2 [d] (C-15), 45.8 [d] (C-8),](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-212-320.jpg)
![DARSTELLUNG DER SUBSTRATE ZUR ENZYMATISCHEN RACEMATSPALTUNG 199
44.8 [d] (C-2), 37.5 [u] (C-17), 36.4 [u] (C-21), 28.8 [u] (C-4), 18.5 [u] (C-18), 18.2 [u]
(C-22), 13.9 [d] (C-19), 13.7 [d] (C-23).
MS (CI, Isobutan, DEP, 100 eV) m/z (%): 477 ([M + C4H9]+, 2), 420 ([M + H]+, 100),
375 (11), 355 (13), 332 (63), 287 (73), 275 (15), 199 (44), 187 (23), 113 (13), 97 (10),
85 (24), 83 (21), 81 (30), 79 (14), 71 (31).
IR (kapillar): ∼ = 2963 (s), 2943 (s), 2874 (m), 2819 (m), 2765 (m), 1733 (s), 1605
ν
(m), 1585 (m), 1490 (m), 1461 (s), 1433 (s), 1382 (m), 1362 (m), 1292 (s), 1256 (s),
1191 (s), 1154 (s), 1097 (s), 1049 (m). 1029 (m), 991 (m), 973 (m),849 (w), 803 (w),
778 (w), 700 (w) cm-1.
Die Synthese von rac-25 erfolgte ebenfalls gemäß AAV 3 bei Raumtemperatur in
THF mit einer Ausbeute von 96 %.
3.3.4 Darstellung der racemischen Ester zur Bestimmung des Umsatzes in
der enzymatischen Transacylierung
3.3.4.1 Darstellung von (±)-(1SR,3RS,4RS)-Essigsäure-4-dimethylamino-
methyl-3-hydroxy-3-(3-methoxy-phenyl)-cyclohexylester (rac-47)
Die Synthese erfolgte gemäß AAV 3 bei Raumtemperatur, dazu wurden 0.070 g
(0.25 mmol) rac-12, gelöst in 1 ml abs. THF, mit 0.072 g (0.64 mmol) Kalium-tert-
butylat und über einen Zeitraum von 10 Min. mit 0.031 g (0.39 mmol)
Essigsäurechlorid, gelöst in 0.01 ml abs. THF, versetzt. Nach Aufarbeitung und
Säulenchromatographie (Diisopropylether : MeOH = 1 : 1 über Kieselgel, Rf = 0.30;
Rf (rac-12) = 0.18) wurden 0.054 g (0.17 mmol, 68 %) eines klaren, gelblichen Öls
erhalten.](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-213-320.jpg)
![200 EXPERIMENTELLER TEIL
O OH
5 10
6 1 9 11
16 O O
17 7 15
3 14
4 2
N 12
13
8
rac-47: MW = 321.4 g⋅mol-1
H-NMR (400 MHz, CDCl3): δ = 7.17 ppm (t, 3J = 8 Hz, 1 H, 13-H), 7.06 (s, 1 H,
1
10-H), 6.95 (d, 3J = 7 Hz, 1 H, 12-H), 6.69 (dd, 3J = 8 Hz, 4J = 3 Hz, 1 H, 14-H), 5.12
(m, 1 H, 5-H), 3.73 (s, 3 H, 15-H), 2.32 (dd, 2J = 14 Hz, 3J = 4 Hz, 1 H, 7-H´), 2.26 –
1.10 (m, 17 H, 2-H, 3-H, 4-H, 6-H, 7-H, 8-H, 17-H).
13
C-NMR (100 MHz, CDCl3): δ = 169.0 ppm [u] (C-16), 158.4 [u] (C-11), 149.1 [u]
(C-9), 127.9 [d] (C-13), 115.9 [d] (C-14), 110.3 [d] (C-12), 109.6 [d] (C-10), 77.0 [u]
(C-1), 69.9 [d] (C-5), 59.4 [u] (C-7), 54.1 [d] (C-15), 46.7 [d] (C-8), 44.9 [u] (C-6), 43.1
[d] (C-2), 31.2 [u] (C-4), 24.8 [u] (C-3), 20.3 [d] (C-17).
MS (EI, 70 eV) m/z (%): 321 (M+, 8), 135 (4), 86 (11), 84 (18), 58 (100), 47 (4).
IR (kapillar): ∼ = 2946 (m), 2830 (w),1730 (s),1601 (w), 1584 (w), 1483 (w), 1462
ν
(m), 1431 (w), 1287 (m), 1250 (s), 1157 (w), 1034 (m), 785 (w), 733 (m) cm-1.
3.3.4.2 Darstellung von (±)-(1SR,3RS,4RS)-Propionsäure-4-dimethylamino-
methyl-3-hydroxy-3-(3-methoxy-phenyl)-cyclohexylester (rac-46)
Die Synthese erfolgte gemäß AAV 3 bei Raumtemperatur, dazu wurden 0.070 g
(0.25 mmol) rac-12, gelöst in 1 ml abs. THF, mit 0.072 g (0.64 mmol) Kalium-tert-
butylat und über einen Zeitraum von 10 Min. mit 0.036 g (0.39 mmol)
Propionsäurechlorid, gelöst in 0.01 ml abs. THF, versetzt. Nach Aufarbeitung und
Säulenchromatographie (Diisopropylether : MeOH = 1 : 1 über Kieselgel, Rf = 0.36;
Rf (rac-12) = 0.18) wurden 0.060 g (0.18 mmol, 71 %) eines klaren, schwach
gelblichen sirupösen Öls erhalten.](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-214-320.jpg)
![DARSTELLUNG DER SUBSTRATE ZUR ENZYMATISCHEN RACEMATSPALTUNG 201
O OH
18 5 10
6 1 9 11
16 O O
17 3 7 15
4 2 14
N 12
13
8
rac-46: MW = 335.4 g⋅mol-1
H-NMR (400 MHz, CDCl3): δ = 7.17 ppm (t, 3J = 8 Hz, 1 H, 13-H), 7.07 (s, 1 H,
1
10-H), 6.96 (d, 3J = 7 Hz, 1 H, 12-H), 6.69 (md, 3J = 8 Hz, 1 H, 14-H), 5.13 (m, 1 H,
5-H), 3.74 (s, 3 H, 15-H), 2.33 (dd, 2J = 14 Hz, 3J = 4 Hz, 1 H, 7-H´), 2.26 – 1.30 (m,
16 H, 2-H, 3-H, 4-H, 6-H, 7-H, 8-H, 17-H), 1.01 (t, 3J = 8 Hz, 3 H, 18-H).
13
C-NMR (100 MHz, CDCl3): δ = 172.7 ppm [u] (C-16), 159.4 [u] (C-11), 150.1 [u]
(C-9), 129.0 [d] (C-13), 117.0 [d] (C-14), 111.5 [d] (C-12), 110.7 [d] (C-10), 78.0 [u]
(C-1), 70.6 [d] (C-5), 60.9 [u] (C-7), 55.5 [d] (C-15), 48.2 [d] (C-8), 46.3 [u] (C-6), 44.5
[d] (C-2), 32.6 [u] (C-4), 38.3 [u] (C-17), 26.3 [u] (C-3), 9.6 [d] (C-18).
MS (EI, 70 eV) m/z (%): 335 (M+, 12), 279 (3), 135 (5), 84 (11), 58 (100).
IR (kapillar): ∼ = 2944 (s), 2862 (m), 2831 (m), 2783 (m), 1732 (s), 1601 (m), 1584
ν
(m), 1483 (m), 1462 (s), 1429 (m), 1351 (w), 1286 (m), 1255 (s), 1193 (s), 1158 (m),
1083 (m), 1048 (m), 1023 (m), 785 (m), 732 (w), 702 (w) cm-1.
3.3.4.3 Darstellung von (±)-(1SR,2RS,4RS)-Essigsäure-3-dimethylamino-
methyl-4-hydroxy-4-(3-methoxy-phenyl)-cyclohexylester (rac-24)
Die Synthese erfolgte, indem 0.070 g (0.25 mmol) rac-14, gelöst in 1 ml
Dichlormethan, mit 2.3 ml (25 mmol) Vinylacetat und 500 mg Novozym 435 versetzt
und bei Raumtemperatur gerührt wurden. Die Rührgeschwindigkeit wurde dabei nicht
zu hoch gewählt, um den immobilisierten Katalysator nicht zu beeinträchtigen. Nach
96 h konnte kein Edukt 14 mehr detektiert werden. Die Reaktionslösung wurde vom
Katalysator abfiltriert, mit 2 ml Dichlormethan versetzt und für weitere 15 h mit 5 ml
ges. NaHCO3-Lösung gerührt, um während der Reaktion entstandene Essigsäure zu
neutralisieren. Nach Phasentrennung wurde dreimal mit je 5 ml Dichlormethan](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-215-320.jpg)
![202 EXPERIMENTELLER TEIL
nachextrahiert. Die vereinigten organischen Phasen wurden über NaSO4 getrocknet.
Nach Einengen am Rotationsverdampfer wurden 0.080 g (0.25 mmol, 99 %)
gelbliches Öl erhalten.
OH
10
5 6 1 9 11
O
17 16 O 3 7 15
4 2 14
N 12
O 8
13
rac-24: MW = 321.4 g⋅mol-1
H-NMR (400 MHz, CDCl3): δ = 7.16 ppm (t, 3J = 8 Hz, 1 H, 13-H), 7.03 (s, 1 H,
1
10-H), 6.91 (s, 1 H, 12-H), 6.68 (dd, 3J = 8 Hz, 4J = 3 Hz, 1 H, 14-H), 4.78 (m, 1 H, 4-
H), 3.71 (s, 3 H, 15-H), 2.33 (dd, 2J = 14 Hz, 3J = 4 Hz, 1 H, 7-H´), 2.20 – 1.55 (m,
17 H, 2-H, 3-H, 5-H, 6-H, 7-H, 8-H, 17-H).
13
C-NMR (100 MHz, CDCl3): δ = 169.4 ppm [u] (C-16), 158.3 [u] (C-11), 149.3 [u]
(C-9), 127.9 [d] (C-13), 116.0 [d] (C-14), 110.5 [d] (C-12), 109.7 [d] (C-10), 74.6 [u]
(C-1), 72.2 [d] (C-4), 59.9 [u] (C-7), 54.0 [d] (C-15), 46.7 [d] (C-8), 41.9 [d] (C-2), 38.0
[u] (C-6), 31.9 [u] (C-3), 26.5 [u] (C-5), 20.4 [d] (C-17).
MS (EI, 70 eV) m/z (%): 322 (3), 321 (M+, 11), 135 (3), 118 (3), 86 (57), 84 (91), 58
(100), 49 (16), 47 (16).
IR (kapillar): ∼ = 2951 (s), 2862 (w), 2831 (m), 2781 (w), 1730 (s), 1602 (s), 1584
ν
(s), 1483 (s), 1463 (s), 1431 (m), 1366 (m), 1317 (w), 1286 (s), 1252 (s), 1171 (m),
1094 (w), 1034 (s), 998 (m), 962 (w), 913 (m), 786 (m), 733 (s), 704 (m) cm-1.](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-216-320.jpg)
![DARSTELLUNG DER SUBSTRATE ZUR ENZYMATISCHEN RACEMATSPALTUNG 203
3.3.4.4 Darstellung von (±)-(1SR,3RS,4RS)-Propionsäure-4-dimethylamino-
methyl-3-hydroxy-3-(3-methoxy-phenyl)-cyclohexylester (rac-45)
Die Synthese erfolgte gemäß AAV 3 bei Raumtemperatur, dazu wurden 0.070 g
(0.25 mmol) rac-14, gelöst in 1 ml abs. THF, mit 0.072 g (0.64 mmol) Kalium-tert-
butylat und über einen Zeitraum von 10 Min. mit 0.036 g (0.39 mmol)
Propionsäurechlorid, gelöst in 0.01 ml abs. THF, versetzt. Nach Aufarbeitung und
Säulenchromatographie (Diisopropylether : MeOH = 1 : 1 über Kieselgel, Rf = 0.33;
Rf (rac-14) = 0.14) wurden 0.055 g (0.16 mmol, 66 %) eines klaren, schwach
gelblichen sirupösen Öls erhalten.
OH
10
5 6 1 9 11
17 O
16 O 3 7 15
4 2 14
18 N 12
O 8
13
rac-45: MW = 335.4 g⋅mol-1
H-NMR (400 MHz, CDCl3): δ = 7.17 ppm (t, 3J = 8 Hz, 1 H, 13-H), 7.05 (s, 1 H,
1
10-H), 6.93 (s, 1 H, 12-H), 6.68 (dd, 3J = 8 Hz, 4J = 3 Hz, 1 H, 14-H), 4.77 (m, 1 H, 4-
H), 3.72 (s, 3 H, 15-H), 2.50 – 1.55 (m, 17 H, 2-H, 3-H, 5-H, 6-H, 7-H, 7-H´, 8-H, 17-
H), 1.07 (t, 3J = 7 Hz, 3 H, 18-H).
13
C-NMR (100 MHz, CDCl3): δ = 174.2 ppm [u] (C-16), 159.7 [u] (C-11), 150.7 [u]
(C-9), 129.3 [d] (C-13), 117.4 [d] (C-14), 111.8 [d] (C-12), 111.0 [d] (C-10), 76.0 [u]
(C-1), 73.4 [d] (C-4), 61.3 [u] (C-7), 55.4 [d] (C-15), 48.1 [d] (C-8), 43.3 [d] (C-2), 39.4
[u] (C-6), 33.2 [u] (C-3), 28.2 [u] (C-17), 27.8 [u] (C-5), 9.5 [d] (C-18).
MS (EI, 70 eV) m/z (%): 335 (M+, 10), 135 (3), 86 (37), 84 (57), 58 (100), 49 (7).
IR (kapillar): ∼ = 2947 (s), 2861 (w), 2831 (m), 2782 (w), 1728 (s), 1602 (s), 1584
ν
(s), 1483 (s), 1728 (s), 1602 (s), 1584 (s), 1483 (s), 1463 (s), 1430 (s), 1352 (w),
1287 (m), 1255 (m), 1193 (s), 1084 (s), 1047 (s), 1020 (s), 997 (m), 962 (w), 911 (w),
785 (m), 734 (s), 704 (m) cm-1.](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-217-320.jpg)
![204 EXPERIMENTELLER TEIL
3.3.5 Versuche zur Darstellung von (±)-(1RS,5RS,8RS)-8-
Dimethylaminomethyl-1-(3-methoxy-phenyl)-2,4-dioxa-bicyclo-[3.3.1]-
nonan-3-on (rac-48)
3.3.5.1 Versuch zur Darstellung von rac-48 durch Acylierung mit Bis-
(trichlormethyl)-carbonat222,246
1.150 g (3.6 mmol) rac-13⋅HCl und 2.888 g (36.5 mmol) Pyridin wurden in 20 ml abs.
Dichlormethan gelöst und auf −76 ºC gekühlt. Zu der klaren Lösung wurde über
einen Zeitraum von 10 Min. bei −76 ºC eine Lösung von 0.520 g (1.75 mmol) Bis-
(trichlormethyl)-carbonatf (Triphosgen) in 8 ml abs. Dichlormethan mittels Spritzen-
technik gegeben. Anschließend wurde noch 1.5 Stunden bei −76 ºC gerührt, dabei
verfärbte sich die Lösung schwach gelb und es bildete sich ein weißer Feststoff.
Nachdem die Lösung über einen Zeitraum von 1 Stunde auf Raumtemperatur
erwärmt wurde, wurde ein Überschuß an ges. NaHCO3-Lösung zugegeben und für
weitere 10 Stunden gerührt. Nach Phasentrennung wurde dreimal mit jeweils 15 ml
Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden über NaSO4
getrocknet, und das Lösungsmittel wurde am Rotationsverdampfer entfernt. Es
konnten 1.284 g einer Rohmischung erhalten werden, welche noch Pyridin enthielt.
Nach Säulenchromatographie (Diisopropylether : MeOH = 1 : 1 über Kieselgel,
Rohmischung: Rf (Pyridin) = 0.68, Rf (1) = 0.34, Rf (2) = 0.28, Rf (rac-13) = 0.10)
konnte allerdings nur eine Mischung von nicht näher charakterisierten
Zersetzungsprodukten und 0.334 g rac-13 erhalten werden.
Ein analoger Versuch mit rac-13 als Edukt führte zum gleichen Ergebnis.
f
S. B. Damle, Safe handling of diphosgene and triphosgene, Chem. & Eng. News 1993, 71, 4.](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-218-320.jpg)

![206 EXPERIMENTELLER TEIL
C-NMR (100 MHz, CDCl3): δ = 159.8 ppm [u] (C-11), 148.4 [u] (C-9), 142.5 [u]
13
(C-1), 137.3 [n. b.] (C-2), 133.1 [u] (C-16), 130.8 [d] (C-17), 129.6 [d] (C-13), 124.1
[d] (C-17/19), 117.8 [d] (C-14), 117.3 [d] (C-17/19), 112.7 [d] (C-12), 111.3 [d] (C-10),
75.2 [d] (C-5), 55.5 [d] (C-15), 33.0 [u] (C-6), 26.9 [u] (C-4), 23.5 [u] (C-3).
Vor Säulenchromatographie: MS (EI, 70 eV) m/z (%): 305 (M+[rac-48], 22), 298
(M+[rac-49], 20), 186 (83), 121 (17), 58 (100).
Ein analoger Versuch mit rac-13⋅HCl als Edukt führte zum gleichen Ergebnis, hier
konnte allerdings neben rac-49 auch rac-13 in geringen Mengen isoliert werden.](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-220-320.jpg)







![216 EXPERIMENTELLER TEIL
3.6.2 Berechnung des E-Werts
Folgende Formeln wurden zur Berechnung der E-Werte benutzt6,106,108:
ln [1 − c(1+ eeP )]
Formel 3.2: E= , für c < 50 %
ln [1− c(1− ee P )]
ln [(1− c)(1− ee S )]
Formel 3.3: E= , für c > 50 %
ln [(1− c)(1+ ee S )]
ee S
Formel 3.4: Umsatz ber =
ee S + ee P
1− ee S ee P (1− ee S )
ln ln (ee + ee )
1 + (eeS /ee P ) S
E= =
P
Formel 3.5:
1 + ee S ee P (1+ ee S )
ln ln
1 + (eeS /ee P ) ee P + ee S )
(mit: c = Umsatz, ee P = ee-Wert des Produkts, ee S = ee-Wert des Substrats)
Alle E-Werte in dieser Arbeit wurden aus dem ee-Wert des Substrats und dem
ee-Wert des Produkts berechnet. Dazu wurde Formel 3.5, die sich aus Formel 3.4
eingesetzt in Formel 3.2 oder Formel 3.3 ergibt, verwendet.6 Von einer Berechnung
des E-Werts aus dem gemessenen Umsatz und nur einem ee-Wert wurde
abgesehen.](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-230-320.jpg)

![218 EXPERIMENTELLER TEIL
Tabelle 3.8: PLE-katalysierte Hydrolyse des Esters rac-15 im wäßrigen
Einphasensystem
Reaktions- ee Ester Ausbeute ee Phenol Ausbeute
E
zeit (+)-15 Ester (+)-15 (−)-8 Phenol (−)-8
2h 62 % 47 % 47 % 49 % 13
3.6.3.3 PLE-katalysierte Hydrolyse des Esters rac-15 im wäßrigen
Einphasensystem zur Darstellung von (+)-15
Die Enzym-katalysierte Hydrolyse erfolgte gemäß AAV 6 bei pH 8.0, dazu wurden
1.120 g (3.5 mmol) des Esters rac-15 in 8 ml tert-Butanol und 72 ml wäßriger
Phosphatpufferlösung (pH 8.0) gelöst und mit 400 µl (4.0 mg) PLE/(NH4)2SO4-
Suspension (PLE, 130 U/mg) versetzt. Die Reaktionslösung war stark getrübt. Die
Reaktion wurde nach 3 Stunden bei einem Umsatz (GC) von ca. 85 % durch
kontinuierliche Extraktion mit Dichlormethan abgebrochen und aufgearbeitet. Nach
Säulenchromatographie (CHCl3 : i-Propanol = 2 : 1 über Kieselgel, Rf ((−)-8) = 0.22;
Rf ((+)-15) = 0.01) konnten 0.560 g (2.25 mmol, 64 %) (−)-8 und 0.113 g (0.35 mmol,
10 %) (+)-15 isoliert werden.
(+)-15: [α ] 20 = + 10.74º (c = 0.7, MeOH)
D
(−)-8: [α ] 20 = − 3.43º (c = 1.5, MeOH)
D
Tabelle 3.9: PLE-katalysierte Hydrolyse des Esters rac-15 im wäßrigen
Einphasensystem
Reaktions- ee Ester Ausbeute ee Phenol Ausbeute
E
zeit (+)-15 Ester (+)-15 (−)-8 Phenol (−)-8
3h ≥ 98 % 10 % 27 % 64 % 7](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-232-320.jpg)




![ENZYM-KATALYSIERTE RACEMATSPALTUNGEN IM WÄßRIGEN UND ORGANISCHEN MEDIUM 223
Tabelle 3.17: CRL-katalysierte Hydrolyse des Esters rac-15 im wäßrigen
Einphasensystem
Reaktionszeit Umsatz (GC) ee Ester (+)-15 ee Phenol (−)-8 E
3h 64 % 98 % 45 % 10
3.6.3.12 CRL-katalysierte Hydrolyse des Esters rac-15 im wäßrig-organischen
Zweiphasensystem mit roher CRL
Die Enzym-katalysierte Hydrolyse erfolgte gemäß AAV 7 bei pH 7.0, dazu wurden
0.316 g (1.0 mmol) des Esters rac-15 in 20 ml MTBE und 20 ml wäßriger
Phosphatpufferlösung (pH 7.0) gelöst und mit 45 mg Candida rugosa Lipase (CRL;
2.4 U/mg) versetzt. Die Reaktionsemulsion war klar und farblos. Die Reaktion wurde
nach 3.25 Stunden bei einem Umsatz (GC) von ca. 51 % durch kontinuierliche
Extraktion mit Dichlormethan abgebrochen und aufgearbeitet. Nach
Säulenchromatographie (EE : MeOH = 3 : 1 über Kieselgel, Rf ((+)-15) = 0.31;
Rf ((−)-8) = 0.10) konnten 0.203 g (0.64 mmol, 64 %) (+)-15 und 0.076 g (0.30 mmol,
30 %) (−)-8 isoliert werden.
(+)-15: [α ] 20 = + 17.90º (c = 1.3, MeOH)
D
(−)-8: [α ] 20 = − 11.45º (c = 2.0, MeOH)
D
Tabelle 3.18: CRL-katalysierte Hydrolyse des Esters rac-15 im wäßrig-organischen
Zweiphasensystem mit roher CRL
Reaktions- ee Ester Ausbeute ee Phenol Ausbeute
E
zeit (+)-15 Ester (+)-15 (−)-8 Alkohol (−)-8
3.25 h 75 % 64 % 85 % 30 % 27](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-237-320.jpg)










![234 EXPERIMENTELLER TEIL
Rf ((−)-18) = 0.32; Rf ((+)-12) = 0.15) konnten 0.082 g (0.23 mmol, 47 %) (−)-18 und
0.042 g (0.15 mmol, 30 %) (+)-12 isoliert werden.
(−)-18: [α ] 20 = − 6.01º (c = 1.50, MeOH)
D
(+)-12: [α ] 20 = + 21.70º (c = 0.80, MeOH)
D
Tabelle 3.32: PLE-katalysierte Hydrolyse des Esters rac-18
Reaktions- ee Ester Ausbeute ee Alkohol Ausbeute
E
zeit (−)-18 Ester (−)-18 (+)-12 Alkohol (+)-12
5.75 h 86 % 47 % 94 % 30 % 89
3.6.8.2 CRL-katalysierte Hydrolyse des Esters rac-18 im wäßrigen
Einphasensystem
Die Enzym-katalysierte Hydrolyse erfolgte gemäß AAV 6 bei pH 7.0, dazu wurden
0.500 g (1.43 mmol) des Esters rac-18 in 4 ml tert-Butanol und 36 ml wäßriger
Phosphatpufferlösung (pH 7.0) gelöst und mit 5 mg Candida rugosa Lipase (CRL;
37 U/mg) versetzt. Die Reaktionslösung war zu Beginn stark getrübt, am Ende der
Reaktionszeit klar und farblos. Die Reaktion wurde nach 4.75 Stunden bei Umsatz
(GC) von ca. 49 % durch kontinuierliche Extraktion mit Dichlormethan abgebrochen
und aufgearbeitet. Nach Säulenchromatographie (Diisopropylether : MeOH = 1 : 1 an
Kieselgel, Rf ((+)-18) = 0.36; Rf ((−)-12) = 0.18) konnten 0.216 g (0.62 mmol, 43 %)
(+)-18 und 0.162 g (0.58 mmol, 41 %) (−)-12 isoliert werden.
(+)-18: [α ] 20 = + 7.03º (c = 0.72, MeOH)
D
(−)-12: [α ] 20 = − 28.30º (c = 1.00, MeOH)
D](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-248-320.jpg)
![ENZYM-KATALYSIERTE RACEMATSPALTUNGEN IM WÄßRIGEN UND ORGANISCHEN MEDIUM 235
Tabelle 3.33: CRL-katalysierte Hydrolyse des Esters rac-18 im wäßrigen
Einphasensystem
Reaktions- ee Ester Ausbeute ee Alkohol Ausbeute
E
zeit (+)-18 Ester (+)-18 (−)-12 Alkohol (−)-12
4.75 h 93 % 41 % 95 % 41 % 133
3.6.8.3 CRL-katalysierte Hydrolyse des Esters rac-18 im wäßrig-organischen
Zweiphasensystem
Die Enzym-katalysierte Hydrolyse erfolgte gemäß AAV 7 bei pH 7.5, dazu wurden
0.349 g (1.0 mmol) des Esters rac-18 in 26 ml MTBE und 26 ml wäßriger
Phosphatpufferlösung (pH 7.5) gelöst und mit 6 mg Candida rugosa Lipase (CRL;
37 U/mg) versetzt. Die Reaktionsemulsion war klar und farblos. Die Reaktion wurde
nach 72 Stunden bei einem Umsatz (GC) von ca. 45 % durch kontinuierliche
Extraktion mit Dichlormethan abgebrochen und aufgearbeitet. Nach
Säulenchromatographie (Diisopropylether : MeOH = 1:1 an Kieselgel,
Rf ((+)-18) = 0.36; Rf ((−)-12) = 0.18) konnten 0.140 g (0.40 mmol, 40 %) (+)-18 und
0.110 g (0.39 mmol, 39 %) (−)-12 isoliert werden.
(+)-18: [α ] 20 = + 7.03º (c = 0.72, MeOH)
D
(−)-12: [α ] 20 = − 28.30º (c = 1.00, MeOH)
D
Tabelle 3.34: CRL-katalysierte Hydrolyse des Esters rac-18 im wäßrig-organischen
Zweiphasensystem
Reaktions- ee Ester Ausbeute ee Alkohol Ausbeute
E
zeit (+)-18 Ester (+)-18 (−)-12 Alkohol (−)-12
72 h ≥98 % 40% ≥98 % 39 % >200](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-249-320.jpg)
![236 EXPERIMENTELLER TEIL
3.6.8.4 CRL-katalysierte Hydrolyse des Esters rac-18 im wäßrig-organischen
Zweiphasensystem mit extraktiver Aufarbeitung
Die Enzym-katalysierte Hydrolyse erfolgte gemäß AAV 7 bei pH 7.2, dazu wurden
0.500 g (1.43 mmol) des Esters rac-18 in 25 ml MTBE und 25 ml wäßriger
Phosphatpufferlösung (pH 7.2) gelöst und mit 13 mg Candida rugosa Lipase (CRL;
25.9 U/mg) versetzt. Nach 48 Stunden bei einem Umsatz von ca. 50 % wurde zum
Abbruch der Reaktion und zur Aufarbeitung die Reaktionslösung zunächst einmal mit
40 ml MTBE extrahiert und anschließend dreimal mit je 30 ml Diethylether. Die
vereinigten organischen Phasen wurden mit Na2SO4 getrocknet und im
Rotationsverdampfer eingeengt. Es konnten 0.254 g (0.73 mmol, 50.8 %) farbloser,
öliger Ester (+)-18 mit einem ee-Wert von ≥98 % isoliert werden. Die verbliebene
wäßrige Phase wurde am Rotationsverdampfer unter vermindertem Druck von
Lösungsmittelresten befreit und durch Zugabe von Na2CO3 auf pH 10 eingestellt.
Anschließend wurde viermal mit je 30 ml Dichlormethan extrahiert und die
vereinigten organischen Phasen über Na2SO4 getrocknet. Nach Einengen am
Rotationsverdampfer wurden 0.184 g (0.66 mmol, 46.1 %) Alkohol (−)-12 mit einem
ee-Wert von 97 % erhalten.
(+)-18: [α ] 20 = + 7.9º (c = 0.69, MeOH)
D
(−)-12: [α ] 20 = − 28.60º (c = 0.93, MeOH)
D
Tabelle 3.35: CRL-katalysierte Hydrolyse des Esters rac-18 wäßrig-organischen
Zweiphasensystem mit extraktiver Aufarbeitung
Reaktions- ee Ester Ausbeute ee Alkohol Ausbeute
E
zeit (+)-18 Ester (+)-18 (−)-12 Alkohol (−)-12
48 h ≥98 % 51 % 97 % 46 % >200](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-250-320.jpg)















![ENZYM-KATALYSIERTE RACEMATSPALTUNGEN IM WÄßRIGEN UND ORGANISCHEN MEDIUM 255
3.6.13.7 CE-katalysierte Hydrolyse des Esters rac-22 im wäßrigen
Einphasensystem mit extraktiver Aufarbeitung
Die Enzym-katalysierte Hydrolyse erfolgte gemäß nach AAV 6 bei pH 7.0, dazu
wurden 0.200 g (0.57 mmol) des Esters rac-22 in 4 ml tert-Butanol und 36 ml
wäßriger Phosphatpufferlösung (pH 7.0) gelöst und mit 15 mg Cholesterolesterase
(CE; 17.5 U/mg) versetzt. Die Reaktionslösung war klar und farblos. Zum Abbruch
der Reaktion nach 22.5 Stunden und zur Aufarbeitung wurde die Reaktionslösung
dreimal mit je 40 ml Diethylether extrahiert. Die vereinigten organischen Phasen
wurden mit Na2SO4 getrocknet und im Rotationsverdampfer eingeengt. Es konnten
0.105 g (0.30 mmol, 53 %) (+)-22 mit einem ee-Wert von 91 % isoliert werden. Die
verbliebene wäßrige Phase wurde am Rotationsverdampfer unter vermindertem
Druck von Lösungsmittelresten befreit und durch Zugabe von Na2CO3 auf pH 10
eingestellt. Anschließend wurde dreimal mit je 30 ml Dichlormethan extrahiert und
die vereinigten organischen Phasen über Na2SO4 getrocknet. Nach Einengen am
Rotationsverdampfer wurden 0.071 g (0.25 mmol, 45 %) (−)-14 mit einem ee-Wert
von 91 % erhalten.
(+)-22: [α ] 20 = + 14.14º (c = 1.02, MeOH)
D
(−)-14: [α ] 20 = − 34.75º (c = 1.05, MeOH)
D
Tabelle 3.63: CE-katalysierten Hydrolyse des Esters rac-22 mit extraktiver
Aufarbeitung
Reaktions- ee Ester Ausbeute ee Alkohol Ausbeute
E
zeit (+)-22 Ester (+)-22 (−)-14 Alkohol (−)-14
22.5 h 91 % 53 % 91 % 45 % 67](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-269-320.jpg)


![258 EXPERIMENTELLER TEIL
3.6.13.11 Chirazyme E1-katalysierte Hydrolyse des Hydrochlorids von Ester
rac-22 (rac-22⋅HCl) im wäßrigen Einphasensystem
Chirazyme E1-katalysierte Hydrolyse des Hydrochlorids von Ester rac-22
(rac-22⋅HCl) unter Zusatz von Aceton als Cosolvens
1.00 g (2.59 mmol) rac-22⋅HCl wurden zu einer Mischung aus 2 ml (16 Vol%) Aceton
und 10.8 ml wäßriger Phosphatpuffer-Lösung (0.1 M, pH 7.0) gegeben. Die Lösung
wurde anschließend auf pH 7.0 eingestellt. Nachdem durch GC-Reaktionskontrolle
überprüft worden war, daß keine nicht-enzymatische Reaktion stattgefunden hatte,
wurden 9 mg Chirazyme E1 (PLE, Fraktion 1, 33.0 U/mg) zugegeben. Danach wurde
der Ansatz bei Raumtemperatur über den gesamten Reaktionszeitraum kräftig
gerührt. Der pH-Wert der Reaktionslösung wurde durch einen pH-STAT Autotitrator
mit 1 N NaOH-Lösung konstant gehalten. Der Verbrauch an NaOH-Lösung wurde
über die gesamte Reaktionsdauer verfolgt. Zur HPLC-Reaktionskontrolle wurden
analog zur AAV 6 zehn 100 µl Proben entnommen. Nach 24 Stunden wurde zum
Abbruch der Reaktion und zur Aufarbeitung die Reaktionslösung zunächst viermal
mit je 30-40 ml MTBE extrahiert. Die vereinigten organischen Phasen wurden mit
Na2SO4 getrocknet und im Rotationsverdampfer eingeengt. Es konnten 0.591 g
(1.69 mmol, 65 %) farbloser, öliger Ester (−)-22 mit einem ee-Wert von 28 % isoliert
werden. Die verbliebene wäßrige Phase wurde am Rotationsverdampfer unter
vermindertem Druck von Lösungsmittelresten befreit und durch Zugabe von Na2CO3
auf pH 10 eingestellt. Anschließend wurde fünfmal mit je 30 ml Dichlormethan
extrahiert und die vereinigten organischen Phasen über Na2SO4 getrocknet. Nach
Einengen am Rotationsverdampfer wurden 0.165 g einer Mischung aus 82 %
(0.46 mmol, 18 %) Alkohol (+)-14 mit einem ee-Wert von 91 % und 18 % (0.10 mmol,
4 %) Ester (−)-22 (mit einem ee-Wert von 28 %) erhalten.
(−)-22: [α ] 20 = − 4.62º (c = 0.96, MeOH)
D
(+)-14: [α ] 20 = + 26.40º (c = 0.32, MeOH)
D](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-272-320.jpg)


![262 EXPERIMENTELLER TEIL
(0.62 mmol, 54 %) durch Spuren von Hexansäure penetrant ziegenähnlich
riechender Ester (−)-23 mit einem ee-Wert von 92 % isoliert werden. Die verbliebene
wäßrige Phase wurde am Rotationsverdampfer unter vermindertem Druck von
Lösungsmittelresten befreit und durch Zugabe von Na2CO3 auf pH 10 eingestellt.
Anschließend wurde fünfmal mit je 30 ml Dichlormethan extrahiert und die
vereinigten organischen Phasen über Na2SO4 getrocknet. Nach Einengen am
Rotationsverdampfer wurden 0.146 g (0.52 mmol, 46 %) (+)-14 als Öl mit einem ee-
Wert von 77 % erhalten.
(−)-23: [α ] 20 = − 6.70º (c = 1.93, MeOH)
D
(+)-14: [α ] 20 = + 25.83º (c = 0.9, MeOH)
D
Tabelle 3.71: Chirazyme E1-katalysierten Hydrolyse des Esters rac-23 unter Zusatz
von Aceton als Cosolvens
Reaktions- ee Ester Ausbeute ee Alkohol Ausbeute
E
zeit (−)-22 Ester (−)-22 (+)-14 Alkohol (+)-14
24 h 92 % 54 % 77 % 46 % 24
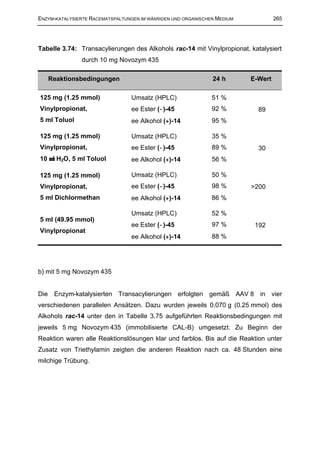
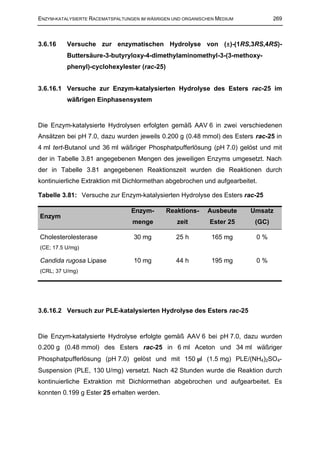
3.6.15 Enzymatische Transacylierungen von (±)-(1RS,2RS,4SR)-2-
Dimethylaminomethyl-1-(3-methoxy-phenyl)-cyclohexan-1,4-diol
(rac-14)
3.6.15.1 CRL-katalysierte Transacylierungen des Alkohols rac-14
Die Enzym-katalysierte Transacylierungen erfolgten gemäß AAV 8 in vier
verschiedenen parallelen Ansätzen. Dazu wurden jeweils 0.070 g (0.25 mmol) des
Alkohols rac-14 unter den in Tabelle 3.72 aufgeführten Reaktionsbedingungen mit
jeweils 5 mg Candida rugosa Lipase (CRL; 37 U/mg) umgesetzt. In allen Reaktionen
entstand eine klare Lösung in der das unlösliche Enzym suspendiert war.](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-276-320.jpg)






![ANHANG ZUM EXPERIMENTELLEN TEIL 271
3.7 Anhang zum experimentellen Teil
3.7.1 Programm zur Bestimmung der Esteraseaktivität von PLE durch
Verseifung von Buttersäureethylester
Mode STAT
Regelparameter Endpunkt pH 8.00
Regelbereich 0.5
Max. Rate 10.0 ml/Min.
Min. Rate 25.0 µl/Min.
Titrationsparameter Start V aus
Start 0s
Start pH aus
Startrate aus
Zeitintervall 60 s
Titrationsrichtung +
Meßeingang 1
Parameter
Abbruchbedingungen Stoppzeit abs. 7000 s
Stoppvolumen abs. 99.99 ml
Stopprate aus
Füllgeschwindigkeit max
Statistik Status aus
Auswertung U. Grenze 1 aus
Fix-V1 bis Fix-V9 alle 600 s (→ C51 bis C59)
Fix-Zeit 1 aus
Überwachung Alle Unterpunkte aus
Vorwahl Ident. abfragen aus
Einmass abfragen alle (→ C00)
Rate anzeigen aus
Aktivierpuls aus
c-fml C01=0.01
Formel RS1 = (C52 − C51) / (C00 ∗ C01) = 10 – 20 Min. [U]
Report Kurve, voll, Liste](https://image.slidesharecdn.com/griebelcarsten-120314145749-phpapp01/85/Enzyme-catalyzed-kinetic-resolution-of-racemic-tramadol-and-its-analogues-industrial-applicability-285-320.jpg)










































