Europe IVD medical registration and approval chart - EMERGO
•
1 gefällt mir•4,694 views

Easy to understand chart describes the IVD regulatory CE Marking process for the European Union.

Empfohlen
Empfohlen
Weitere ähnliche Inhalte
Was ist angesagt?
Was ist angesagt? (20)
Andere mochten auch
Andere mochten auch (13)
Ähnlich wie Europe IVD medical registration and approval chart - EMERGO
Ähnlich wie Europe IVD medical registration and approval chart - EMERGO (20)
Kürzlich hochgeladen
Kürzlich hochgeladen (20)
Europe IVD medical registration and approval chart - EMERGO
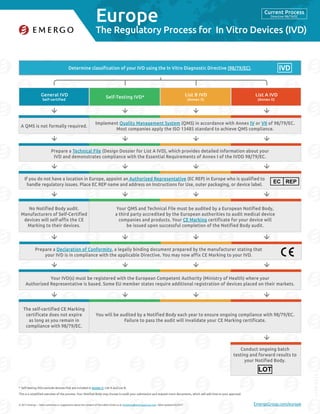
- 1. Europe © 2017 Emergo – Have comments or suggestions about the content of this table? Email us at marketing@emergogroup.com. Table updated 02/2017. EmergoGroup.com/europe Determine classification of your IVD using the In Vitro Diagnostic Directive (98/79/EC). General IVD Self-certified Self-Testing IVD* List B IVD (Annex II) List A IVD (Annex II) 5175-0217 The Regulatory Process for In Vitro Devices (IVD) Self-testing IVDs exclude devices that are included in Annex II, List A and List B. This is a simplified overview of the process. Your Notified Body may choose to audit your submission and request more documents, which will add time to your approval. Implement Quality Management System (QMS) in accordance with Annex IV or VII of 98/79/EC. Most companies apply the ISO 13485 standard to achieve QMS compliance. A QMS is not formally required. Your QMS and Technical File must be audited by a European Notified Body, a third party accredited by the European authorities to audit medical device companies and products. Your CE Marking certificate for your device will be issued upon successful completion of the Notified Body audit. No Notified Body audit. Manufacturers of Self-Certified devices will self-affix the CE Marking to their devices. You will be audited by a Notified Body each year to ensure ongoing compliance with 98/79/EC. Failure to pass the audit will invalidate your CE Marking certificate. The self-certified CE Marking certificate does not expire as long as you remain in compliance with 98/79/EC. Prepare a Technical File (Design Dossier for List A IVD), which provides detailed information about your IVD and demonstrates compliance with the Essential Requirements of Annex I of the IVDD 98/79/EC. Your IVD(s) must be registered with the European Competent Authority (Ministry of Health) where your Authorized Representative is based. Some EU member states require additional registration of devices placed on their markets. If you do not have a location in Europe, appoint an Authorized Representative (EC REP) in Europe who is qualified to handle regulatory issues. Place EC REP name and address on Instructions for Use, outer packaging, or device label. EC REP Prepare a Declaration of Conformity, a legally binding document prepared by the manufacturer stating that your IVD is in compliance with the applicable Directive. You may now affix CE Marking to your IVD. * IVD Conduct ongoing batch testing and forward results to your Notified Body. LOT Current Process Directive 98/79/EC
- 2. Europe Notes 1. The time frames shown above are typical for the majority of IVD submissions but assume that your device does not contain animal tissue, medicinal substances or employ entirely novel technology. Your length of approval will depend on the quality and completeness of your technical documentation and how much time you take to address additional information requests from authorities after submission. YOUR SUBMISSION(S) MAY TAKE MORE TIME THAN WHAT IS SHOWN ABOVE. 2. Registrations remain valid for the time specified as long as you do not make changes to your IVD, intended use or indications for use. 3. We recommend starting the re-registration process no later than the time period specified above. However, please consult with your distributor or regulatory expert well before this suggested time to avoid any lapse in your registration. 4. Our rating of the complexity of the registration process is based on our experience and the opinion of nearly 1,000 QA/RA professionals worldwide who were asked to rate the difficulty of registering a device in each country. The European CE Marking process is considered the mid-point to which all other markets are compared. 5. Prices in US Dollars for a single IVD device. 1 = Less than $5,000; 2 = $10,000 - $15,000; 3 = $15,000 - $30,000; 4 = $30,000 - $50,000; 5 = $50,000 or more. Estimated cost includes registration application fees, in-country representation, Notified Body audit fees, submission preparation consulting and translation of documents, if required. Costs assume you already have approval for your device in the United States, Canada, Australia, or Japan. Costs do NOT include product testing, clinical trials or ISO 13485 implementation, if applicable. These costs are generally spread across many markets and thus not specifically attributed to a European CE submission. EmergoGroup.com/europe 5175-0217 © 2017 Emergo – Have comments or suggestions about the content of this table? Email us at marketing@emergogroup.com. Table updated 02/2017. Time, Cost, and Complexity of IVD Registration Self-testing IVDs exclude devices that are included in Annex II, List A and List B. This is a simplified overview of the process. Your Notified Body may choose to audit your submission and request more documents, which will add time to your approval. * Device classification in Europe General IVD Self-certified Self-Testing IVD* List B IVD (Annex II) List A IVD (Annex II) How long you should expect to wait after submission until approval is granted.1 Not applicable 3-5 months 3-5 months 9-12 months Validity period for CE Marking certificate.2 Not applicable 3 years 3 years 3 years Registration renewal should be started this far in advance.3 Not applicable 2 months 2 months 2 months Complexity of the registration process for this classification.4 Overall cost of gaining regulatory approval.5 Simple Complex Simple Complex Simple Complex Simple Complex Low High Low High Low High Low High 1 1 2 3 3 4 44 Current Process Directive 98/79/EC