Tumor de wilms
•Als PPTX, PDF herunterladen•
17 gefällt mir•20,141 views

Presentación para la clase de Nefrología de la Facultad de Medicina, Mexicali UABC.

Empfohlen
Weitere ähnliche Inhalte
Was ist angesagt?
Was ist angesagt? (20)
Ähnlich wie Tumor de wilms
Ähnlich wie Tumor de wilms (20)
Mehr von Elda Soto
Mehr von Elda Soto (10)
Kürzlich hochgeladen
Kürzlich hochgeladen (20)
Tumor de wilms
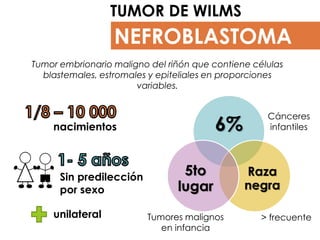
- 1. NEFROBLASTOMA TUMOR DE WILMS Tumor embrionario maligno del riñón que contiene células blastemales, estromales y epiteliales en proporciones variables. 6% Raza negra 5to lugar nacimientos Sin predilección por sexo Cánceres infantiles Tumores malignos en infancia unilateral > frecuente
- 2. ETIOLOGÍA 10% asociado a anomalías congénitas/síndromes 99% esporádico 1% hereditario, autosómico dominante Anomalías congénitas Aniridia Hemihipertrofia Síndromes Beckwith- Wiedemann Denys-Drash WAGR Perlman Golabi-Behmel
- 3. CUADRO CLÍNICO masa abdominal asintomática. Otros hallazgos: Hipertensión (75%) Fiebre (30%) Hematuria microscópica (24%) Dolor (24%) Hematuria macroscópica (18%) Náuseas, vómitos, disminución del apetito, pérdida de peso y constipación. Tamaño de la masa, ascitis, aniridia, extremidades y genitales.
- 4. HISTOPATOLOGÍA Blastemal Estromal Epitelial Neoplasia embrionaria trifásica: Islas de células indiferenciadas. Constituyen estructuras glomeruloides. mixto Factor pronóstico desfavorable ANAPLASIA > 2 años Figuras mitóticas anormales. Núcleos hipercromáticos. Núcleo de diámetro al menos 3 veces > que el de núcleos de células normales.
- 5. HISTOPATOLOGÍA • Grandes, multilobulados • Color gris o bronceado • Áreas focales de hemorragia y necrosis. Características macroscópicas
- 7. ESTADIFICACIÓN I II III IV V Determinante importante en el pronóstico
- 8. ESTADIO I Tumor limitado al riñón y completamente extirpado. Superficie de la cápsula renal está intacta. Tumor no se rompe antes ni durante la escisión. Los vasos del seno renal no están infiltrados. No hay tumor residual más allá de los márgenes de resección. ESTADIFICACIÓN
- 9. ESTADIO II El tumor se extiende más allá del riñón, pero ha sido completamente removido. Tumor penetra la cápsula renal hacia tejidos blandos perirrenales. Compromiso de vasos del seno renal. ESTADIFICACIÓN No hay tumor residual aparente más allá de los márgenes de resección.
- 10. ESTADIFICACIÓN ESTADIO III Tumor residual limitado al abdomen, sin diseminación hematógena. Tumor residual en abdomen. Tumor en ganglios linfáticos abdominales. Contaminación peritoneal difusa/implantes periotoneales. Márgenes quirúrgicos positivos.
- 11. ESTADIFICACIÓN ESTADIO IV Metástasis hematógenas o a ganglios linfáticos extra- abdominales. PULMÓN HÍGADOHUESOS CEREBRO ESTADIO V Compromiso bilateral
- 12. DIAGNÓSTICO Historia clínica y exploración física • Radiografía de tórax • TAC • Ultrasonido adominal Estudios de gabinete • EGO, PFR, PFH, BH. Exámenes de laboratorio • Tumor muy grande para resección primaria segura (inoperables). • Quimioterapia preoperatoria. Biopsia • Diámetro máximo ocupa 2/3 o > de la cavidad abdominal. • Tumor en vena cava • Tumor bilateral.
- 13. TRATAMIENTO Multidisci- plinario Cirugía Quimioterapia Radiación Estadio Volumen tumoral ESTADIOS I II Cirugía + Quimioterapia III IV Biopsia + Quimioterapia + Cirugía + Quimioterapia/Radioterapia V Quimioterapia + Cirugía + Quimioterapia/Radioterapia • 90% en pacientes con tumores localizados. • 70% en pacientes con enfermedad metastásica.
- 14. 1. American Cancer Society. (2015). Wilms Tumor. Recuperado de http://www.cancer.org/cancer/wilmstumor/detailedguide/wilms-tumor- staging 2. Atlas of Pathology. (2009). Nephroblastoma (Wilms Tumor). Recuperado de http://www.pathologyatlas.ro/nephroblastoma-wilms-tumor.php 3. Guía de Práctica Clínica. Tumor de Wilms diagnóstico y tratamiento en pediatría. Recuperado de http://www.cenetec.salud.gob.mx/descargas/gpc/CatalogoMaestro/304_SS A_10_Tumor_Wilms/GRR_SSA_304_10.pdf 4. Paulino, A. (2014). Wilms Tumor. Recuperado de http://emedicine.medscape.com/article/989398-overview 5. Rojas, E. (s.f.). Oncología Pediátrica Tumor de Wilms. Recuperado de http://www.binasss.sa.cr/revistas/rmcc/599/art23.pdf REFERENCIAS
Hinweis der Redaktion
- Neoplasia maligna del riñón, que contiene células blastemales (células indiferenciadas), epiteliales y estromales (constituye ciertos tipos de tejido conjuntivo). Está bien encapsulado en las primeras fases, aunque más tarde se puede extender a los ganglios linfáticos y a la vena renal o cava y metastizar en los pulmones o en otras localizaciones. La incidencia anual se estima en alrededor de 1/8 - 10.000 nacimientos. Afecta principalmente a niños pequeños, entre 1 y 5 años, con un pico máximo de edad a los 3 – 4 años y afecta por igual a ambos géneros. Más del 90% de los tumores de Wilms son unilaterales y 7% es bilateral afectando con mayor frecuencia a niñas. Es el tumor renal maligno más frecuente en niños y ocupa el 5º lugar en frecuencia tras leucemias, linfomas, tumores cerebrales y neuroblastomas y representa alrededor del 6 por ciento de los cánceres infantiles. Son más frecuentes en afroamericanos y menos frecuentes en asiáticos.
- El 99% de los casos de nefroblastoma son esporádicos y, entre estos, el 10% están asociados a anomalías congénitas (aniridia, hemihipertrofia, defectos genitourinarios) o forman parte de síndromes específicos (síndrome de Beckwith-Wiedemann, Denys-Drash, WAGR o Perlman). 1% se debe a causas hereditarias, comportándose de forma autosómico dominante donde los genes alterados son WT1 y WT2. Los niños con el síndrome de Beckwith-Wiedemann corren un riesgo mayor de desarrollar el tumor de Wilms. Aproximadamente, un quinto de los pacientes lo desarrollan. El acrónimo WAGR está conformado por las iniciales de las cuatro enfermedades presentes en el síndrome de WAGR: tumor de Wilms, aniridia, malformaciones genitourinarias y retraso mental. El síndrome de Denys-Drash cursa con seudohermafroditismo masculino, insuficiencia renal de inicio precoz por esclerosis mesangial y un riesgo elevado de tumor de Wilms.
- El 50% de estos tumores se reconocen en la infancia como una masa abdominal asintomática, homogénea, firme y fija, usualmente referida por el familiar al estar bañando o vistiendo al niño. Otros hallazgos son hematuria macroscópica (18%), hematuria microscópica (24%), fiebre (30%), hipertensión (75%), ésta última a causa del aumento de la secreción de renina, por el tumor mismo o por desplazamiento del riñón o la arteria renal y dolor (24%). Puede ir acompañado de náuseas, vómitos, disminución del apetito, pérdida de peso y constipación. Los tumores también pueden son propensos a invadir la vena renal y formar trombos en la vena cava inferior. En el examen físico se deben buscar intencionadamente aniridia (parcial o completa), asimetría facial y de las extremidades, la localización y tamaño de la masa abdominal, que característicamente ocupa la fosa renal, la presencia de ascitis y anormalidades de los genitales.
- Se considera una neoplasia embrionaria trifásica ya que contiene elementos de distintas estirpes histológicas mostrando componentes blastemal, estromal y epitelial. Al microscopio la imagen más característica para su diagnóstico es la formación de un remolino de células nefrogénicas formando un patrón túbulo-glomerular. En un 90% se encuentra un patrón mixto, siendo también comunes los patrones bifásico y menos frecuente los tumores en que predomina o solo se encuentra un tipo celular. Imagen.- Se observa un componente epitelial (túbulos y glomérulos abortivos) rodeado de blastema metanéfrico y células en forma de huso del estroma. El estroma puede incluir músculo, cartílago, hueso, tejido adiposo diferenciado o elementos anaplásicos. En la parte derecha de la imagen se observa como el tumor comprime el parénquima renal normal. La presencia de anaplasia es un factor de pronóstico desfavorable y un marcador de resistencia a la quimioterapia que se presenta entre el 5 al 10% de los tumores, y principalmente en niños mayores a 2 años de edad. La anaplasia puede ser focal o difusa. Entendiéndose como focal a los cambios nucleares que estén estrictamente confinados a una región específica del tumor primario y ausentes de las estructuras que rodean a la lesión, cualquier caso con anaplasia fuera de la lesión primaria es designada como anaplasia difusa.
- Los determinantes más importantes del pronóstico en los niños con tumor de Wilms son la histopatología y el estadio tumoral. La estadificación por el grupo de estudio del tumor de Wilms va del estadio I al V y se basa fundamentalmente en los hallazgos quirúrgicos y anatomopatológicos.
- Del 40 – 45% de los tumores de Wilms son estadio I. En el estadio I, el tumor se puede extraer completamente durante la cirugía y se presentan todas las situaciones siguientes: No se abrió la capa exterior del riñón (cápsula renal intacta) Se encontró el cáncer solamente en el riñón y el tumor no se diseminó a los vasos sanguíneos del riñón. No se encontraron células cancerosas en los bordes del área donde se extirpó el tumor.
- Del 20% de los tumores de Wilms son estadio II. Hay extensión regional del tumor (penetración a través de la superficie externa de la cápsula renal hacia los tejidos blandos perirrenales). Los vasos que se encuentran fuera de la substancia renal están infiltrados o contienen trombos tumorales. No hay tumor residual aparente en los márgenes de la escisión o más allá de ellos.
- Del 20 - 25% de los tumores de Wilms son estadio III. Esta etapa se refiere a que el tumor de Wilms no fue removido completamente y existe un tumor residual limitado al abdomen. El cáncer se ha diseminado a ganglios linfáticos abdominales. Hay contaminación peritoneal difusa por crecimiento tumoral directo o debido a derramamiento del tumor más allá del flanco antes o durante la cirugía. El tumor se extiende más allá de los márgenes quirúrgicos ya sea microscópica o macroscópicamente. El tumor no es completamente resecable debido a la infiltración local a estructuras vitales.
- El 10% de los tumores de Wilms son estadio IV. Metástasis hematógena, al pulmón, hígado con mayor frecuencia, y hueso, cerebro, otros órganos.
- Al realizar la historia clínica se debe investigar si hay antecedentes familiares de cáncer o anomalías congénitas, especialmente de genitales o del sistema urinario. Los estudios de imagen no pueden distinguir de manera fiable un tumor Wilms de uno no Wilms. Sin embargo, pueden sugerir el diagnóstico y ayudan a descartar de acuerdo al patrón de imagen de otras neoplasias menos frecuentes. Estudios de gabinete.- TC de Tórax, está indicada en todos los casos para la búsqueda de metástasis pulmonares. TC Abdominal proporciona información sobre las características de la masa renal, la extensión anatómica del tumor, la presencia de un riñón contralateral con aspecto normal y la posibilidad de afectación bilateral. Ultrasonido proporciona información útil y sobre la consistencia de la mas, la posible afectación intravascular y demostrar la presencia de un riñón contralateral e identificar la extensión del tumor, útil para valorar la presencia de compromiso de la vena renal, o cava, y además del hígado y otras estructuras abdominales, por lo que es un estudio complementario a la TC de abdomen. Exámenes de laboratorio.- encaminados a conocer la función renal, hepática y la condición hematológica basales. Biopsia.- No es una indicación de rutina, debe limitarse a los casos de tumores inoperables al diagnóstico o en aquellos casos en que la condición del paciente sea indicativa de quimioterapia preoperatoria. Tumor inoperable (diámetro máximo del tumor ocupa 2/3 o más de la cavidad abdominal, presencia de tumor en vena cava (como trombo o como invasión a la pared vascular), HTA incontrolada y TW bilateral.
- Incluye quimioterapia, cirugía y radioterapia. La nefrectomía es actualmente el tratamiento primario recomendado para la mayoría de los niños con TW. El tratamiento del tumor de Wilms se basa en dos fases: cirugía seguida de quimioterapia o quimioterapia seguida de cirugía, la cual dependerá del estadio de la enfermedad al momento del diagnóstico Estadios I y II: Se inicia con la cirugía, posteriormente puede continuarse con quimioterapia. Estadios III y IV: Se realiza biopsia si el tamaño del tumor es muy grande. Posteriormente se inicia con quimioterapia preoperatoria para disminuir el tamaño del tumor durante 6 semanas. Se realiza la cirugía del tumor residual y se continúa con quimioterapia y/o radioterapia. Estadio V: No se recomienda realizar biopsia. Se inicia con quimioterapia preoperatorio para reducir el tamaño de la masa. Se realiza nefrectomía parcial o radical, seguida de quimioterapia y/o radioterapia. La radioterapia se reserva para las formas más extendidas o de histología menos favorable. Se observa mayor propensión a presentar insuficiencia renal. En general, el tratamiento preoperatorio se considera para los tumores de Wilms que no se pueden resecar por completo en la operación inicial, por ejemplo, en los casos bilaterales, los que presentan extensión en la cava y la aurícula, y los que ya han producido metástasis. Si no hay suficiente tejido funcional después de la cirugía, el paciente necesitará diálisis. Si no hay evidencia de cáncer dentro de 1 a 2 años, se puede realizar un trasplante renal. En la mayoría de los casos el pronóstico es favorable: la supervivencia es superior al 90%. La énfasis en las manifestaciones clínicas antes mencionadas, en las cuales se debe tener un alto grado de sospecha, y la implementación de los estudios radiológicos adecuados para su diagnóstico, rápido y acertado, el paciente tiene grandes posibilidades de sobrevida si se diagnostica a tiempo.