LA MEDICINA GRECORROMANA HIPOCRATES, HEROFILO Y GALENO
Resumen Legendario Bioquimica - Aminoacidos 2016
1. RESUMEN LEGENDARIO BIOQUIMICA – AMINOACIDOS MIJAIL JN
PRIMERA PARTE: OXIDACION DE AMINOACIDOS Y PRODUCCION DE UREA
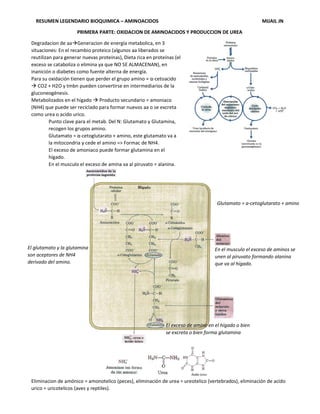
Degradacion de aaGeneracion de energía metabolica, en 3
situaciones: En el recambio proteico (algunos aa liberados se
reutilizan para generar nuevas proteínas), Dieta rica en proteínas (el
exceso se cataboliza o elimina ya que NO SE ALMACENAN), en
inanición o diabetes como fuente alterna de energía.
Para su oxidación tienen que perder el grupo amino = α-cetoacido
CO2 + H2O y tmbn pueden convertirse en intermediarios de la
gluconeogénesis.
Metabolizados en el hígado Producto secundario = amoniaco
(NH4) que puede ser reciclado para formar nuevos aa o se excreta
como urea o acido urico.
Punto clave para el metab. Del N: Glutamato y Glutamina,
recogen los grupos amino.
Glutamato = α-cetoglutarato + amino, este glutamato va a
la mitocondria y cede el amino => Formac de NH4.
El exceso de amoniaco puede formar glutamina en el
hígado.
En el musculo el exceso de amina va al piruvato = alanina.
Eliminacion de amónico = amonotelico (peces), eliminación de urea = ureotelico (vertebrados), eliminación de acido
urico = uricotelicos (aves y reptiles).
El glutamato y la glutamina
son aceptores de NH4
derivado del amino.
Glutamato = a-cetoglutarato + amino
El exceso de amino en el hígado o bien
se excreta o bien forma glutamina
En el musculo el exceso de aminos se
unen al piruvato formando alanina
que va al hígado.
2. Las enzimas se secretan como zimógenos para proteger a la celula que los secreta evitando que la digeran.
Inhibidor pancreático de la tripsina evita la activación prematura de los zimógenos, si se activan = Pancreatitis aguda
(por obstrucción en la ampolla de váter, las enzimas se activan en el páncreas detruyendolo).
DATO: Prots globulares hidrolisis casi completa, prots fibrosas (queratina) hidrolisis parcial.
Consumo diario 50-100g de prots, degradación de 300-400g, resintesis de 300 a 400g y excreción/catabolismo de
50-100 => Balance 0.
ENDOPEPSIDASAS EXOPEPSIDASAS
Pepsina, tripsina, quimotripsina,
elastasa
Aminopepsidasa,
carboxipepsidasas.
1) Ingestion de proteínas
estimula la gastrina.
2) Gastrina Celula parietal = HCl
Celula principal = pepsionogeno
3) Jugo gástrico = pH 2.5,
desnaturaliza las proteínas
ingeridas.
Pepsinogeno+HClPepsina
4)La pepsina hidroliza enlace
peptídico en el amino-terminal
(Phe, Trp, Tyr)
5) Al pasar el contenido al
duodeno, el bajo pH Secretina
6) Secretina Pancreas = HCO3
=> pH A 7.
7) Paralelamente liberación de
colecistoquinina = secreción de
enzimas pancreáticas 1ro el
tripsinogeno que por la
enteropeptidasa = Tripsina
(Hidroliza a mas tripsinogeno y al
resto de zimógenos).
Enzimas pancreáticas:
Tripsina + Quimiotripsina =
Hidrolisis de péptidos que vienen
del estomago.
Carboxipeptidasa A y B =
eliminación de residuo carboxilo –
terminal.
Aminopeptidasa = hidrolisis de
amino-terminal en péptidos cortos.
8) Absorcion de aa por la
vellosidad y al hígado.
3. ENZIMA NIVEL DE ACCION
Pepsina Phe-Phe, Phe-Tyr, Phe-Leu, Leu-
Ala, Leu-Glu
Tripsina Arg-, Lys-
Quimiotripsina Tyr, Phe, Trp, Met, Lys
Elastasa Enlace peptidico
Carboxipepsidasa A Aminoácidos aromáticos
(Phe, Tyr, Trp)
Carboxipepsidasa B Aminoacidos básicos
(Lys, Arg)
Dipeptidasa Dipeptidos
Tripeptidasa Tripeptidos
TRANSPORTE DE AA EN MEMBRANA
Sistema A: Para aa de cadena corta (Ala, gli, ser, met, pro), depende del Na.
Sistema ASC: Para aa neutros (Ala, ser, cis), especifico, depende del Na.
Sistema X: Para aa = Glu y Asp.
Sistema Y: Para aa básicos (Lis, arg), depende del Na.
Sistema N: Especifico para glutamina y asparagina.
Sistema B: Para B-alanina y taurina.
SINDROMES DE MALABSORCION
Cistinuria: Transporte defectuoso de cistina y de aminoácidos dibásicos (arginina, ornitina, y lisina) en la
membrana apical del epitelio intestinal y túbulo proximal renal. Ausencia de reabsorción de cistina en el
túbulo proximal renal produciendo un exceso de cistina en orina y con la consiguiente formación de cálculos
renales. Los cálculos de cistina son muy difíciles de eliminar por litotricia a diferencia del resto de cálculos.
Iminoglicinuria: Consecuencia de un defecto en la reabsorción de la prolina, la hidroxiprolina y la glicina en
el tubo renal. La prevalencia estimada es de 1 de cada 15.000. Normalmente es asintomático y se identifica
de forma fortuita por la detección de niveles elevados de ácidos imino y de glicina en la orina. Se transmite
de forma autosómica recesiva.
Enfermedad de Hartnup: Trastorno metabólico hereditario caracterizado por deficiencias en el mecanismo
de transporte de determinados aminoácidos como el triptófano y la histidina a través del intestino delgado y
los riñones, afeccion mas común de deficiencia en absorción de aminoácidos.
Sintomas: Fotosensibilidad, ataxia cerebelosa, retraso mental, hiperaminoacidemia, dermatitis, cambios en
el estado de ánimo e hipotonía.
TRANSAMINACION
AA en el hígado Eliminacion de α-amino = TRANSAMINACION => Paso del amino desde el aminoácido entrante al
α-cetoglutarato formando además un α-cetoacido, es una reacción REVERSIBLE.
DATO: Prolina, hidroxiprolina, lisina y treonina no participan en transaminacion.
Aminacion de α-cetoglutarato
quedando como glutamato.
Desaminacion de α-aminacido
quedando como α-cetoacido.
Dador de aminos en rutas
biosinteticas y de excreción.
TRANSAMINACION => Enzima = Transaminasa
Cofactor = Piridoxal fosfato (de vit. B6).
Transportador de aminos
Racemizacion (Cα)
Descarboxilacion
Transaminasa = Amino transferasa
4. TRANSAMINASAS Y LESIONES TISULARES
Alanina aminotransferasa (ALT) = Glutamato – piruvato transaminasa (GPT), específicamente
hepática, menor en hepatitis vírica pero mayor en cirrosis, enfermedad hepática, tumores hepáticos,
colestasis y hepatitis toxica.
glutamato + piruvato ⇌ α-cetoglutarato + alanina
Aspartato aminotransferasa (AST) = Glutamato – oxalacetato transaminasa (GOT) en corazón,
hígado y musculo; en cantidades elevadas en infarto agudo de miocardio, hepatopatía y miopatías
(daño celular grave).
L-aspartato + 2-oxoglutarato ⇌ oxalacetato + L-glutamato
Creatina quinasa => Primera en liberarse al suero sanguíneo tras un ataque cardiaco, ellas dan
información sobre la gravedad de la lesión.
Creatina quinasa GOT GTP
DESAMINACION OXIDATIVA
El glutamato recoge los amino, estos deben de eliminarse del glutamato para poder eliminarse.
Citosol Mitocondria Desanimación => Enzima = glutamato deshidrogenasa.
ENZIMA = GLUTAMATO DESHIDROGENASA.
Glutamato deshidrogenasa enzima alosterica: GTP = ATP => No, para no incorporar el α-cetoglutarato a
Krebs y producir mas energia; ADP => Si, se necesita energía.
Glutamato
Deshidrogenasa
Al ciclo de Krebs
5. DESAMINACION NO OXIDATIVA
Para aa alifáticos hidroxilados = Ser y Tre; cofactor = Fosfato de piridoxal. Reaacion de transposición del O del
carbono alfa y beta + eliminación del NH4.
TOXICIDAD DEL AMONIACO
En fases terminales de intoxicación por amoniaco => Edema cerebral + mayor presión intracraneal + agotamiento de
ATP en la neurona. VN: 5-10 uM de amoniaco serico.
En el cerebro producido de degradación de nucleótidos.
Condicion Normal: Liberacion al citosol del exceso de amoniaco => Aminacion del α-cetoglutarato a
glutamato por la glutamato deshidrogenasa + Conversion del glutamato a glutamina por la glutamina
sintetasa.
Glutamato deshidrogenasa + glutamina sintetasa => Altos niveles en el cerebro por tanto si mas amoniaco
entonces mas accion de estas enzimas = mas glutamina (osmolito -> soluto osmóticamente activo) =
captación de agua = edema cerebral => COMA. Adicionalmente un agotamiento del glutamato
(neurotransmisor).
TRANSPORTE DE AMONIACO COMO GLUTAMINA
El amoniaco es muy toxico por tanto niveles regulados, en el transporte del amoniaco al hígado interviene la
GLUTAMINA que se combina con el amoniaco en una reacción catalizada por la glutamina sintetasa.
Glutamina = fuente de aminos en reacciones de biosíntesis.
Glutamina excesiva transportada al intestino, hígado y riñon para ser tratada (eliminación del NH4 y
formación de la urea).
6. En acidosis metabolica, el riñon aumenta la modificación de la glutamina = exceso de NH4 que se elimina
en la orina en forma de sales + descarboxilacion del α-cetoglutarato = HCO3 para el equilibrio acido-basico.
RECORDAR:
TRANSAMINACION (REACCION GLOBAL)
CICLO DE LA GLUCOSA – ALANINA
AA degradados en el musculo, su grupo amino recogido por el glutamato glutamina (transaminacion) / α-
cetoglutarato (alanina aminotransferasa) => alanina va al hígado y ahí se transfiere el amino de la alanina al α-
cetoglutarato formando piruvato + glutamato (entra a la mitocondria y por la DH libera NH4).
Forma de usar el piruvato y lactato (del musculo que no puede hacer gluconeogénesis al hígado que si).
1) Fosforilacion del glutamato
2) Acoplamiento del amoniaco
al glutamil fosfato
3)Accion de la glutaminasa
para formar glutamato y
liberar el NH4
4) El NH4 liberado entra al
ciclo de la urea.
GLUTAMATO
DESHIDROGENASA
REACCION DE DESAMINACION OXIDATIVA
El NH4 liberado entra al ciclo
de la urea.
A
B
7. RECORDAR:
Eliminacion de amónico = amonotelico (peces), eliminación de urea = ureotelico (vertebrados y tiburones),
eliminación de acido urico = uricotelicos (aves y reptiles).
C
8. CICLO DE LA UREA
Localizacion: Mitocondria del HEPATOCITO (inicio) – 3 pasos posteriores en el citosol del HEPATOCITO.
A
B
C
El 1er amino de la matriz
CO2 en forma de bicarbonato
0.1) Accion de la carbamil fosfato
sintasa I = Carbamil fosfato
0.2) El carbamil fosfato => dador de
carbamilo
1) Carbamil fosfato + ornitina -> citrulina
Enzima = ornitina transcarbamilasa
2) La citrulina formada sale de
la mitocondria al citosol
El 2do amino de del aspartato
3) Activacion de
la citrulina por
accion de
arginosuccinato
sintasa (Gasto
de 1 ATP) =
Citrulil AMP
4) Reaccion del
citrulil AMP con el
aspartato por accion
de la arginosuccinato
sintasa formando
arginosuccinato
5) Corte del
arginosuccinato por
accion de
argininosuccinasa =
Arginina + Fumarato
Hacia el ciclo de Krebs
DATO: UNICA REAX.
REVERSIBLE!!!!
6) Accion de la arginasa =
Urea + Ornitina
La ornitina retorna a la matriz
para reiniciar el ciclo, es el
último producto pero es el
primero que reacciona.
9. Sintesis del carbamil fosfato:
Sintesis del arginosuccinato:
Urea: Producto terminal del metabolismo de proteínas, compuesto organico sin enlace C-C o C-H,
hidrosoluble, apolar, difusible, atoxica, 50% de su M es nitrógeno, VN=25-30 gr/dia.
Conexión con el ciclo de Krebs (acido cítrico):
El aspartato del ciclo de Krebs al reaccionar con el citrulil AMP forma argininosuccinato por accion de la
argininosuccinato sintasa que luego por accion de la argininosuccinasa el argininosuccinato = Fumarato +
Argnina, el fumarato producido puede convertirse a malato que es transportado a la mitocondria y ahí entra
al ciclo de Krebs.
Balance Energetico:
Sintesis del carbamil fosfato = 2 ATP
Activacion de la citrulina e ingreso del aspartato = 2 ATP
GASTO: 4 ATP, pero como el malato ingresa a la mitocondria y pasa a Krebs = produce 1 NADH (2.5
ATP).
TOTAL = 4 ATP – 2.5 ATP = 1.5 ATP
Regulacion:
A LARGO PLAZO dependiente del flujo de nitrógeno consumo excesivo de proteínas (eliminación del
exceso) o inanición (degradación de proteína es energía) = mayor producción de urea = mayor síntesis de
enzimas; si consumo muy bajo de proteínas y mas CH = menor síntesis de enzimas del ciclo de la urea.
A CORTO PLAZO por regulación alosterica, paso limitante = reacción de la N-acetilglutamato sintasa que
forma el N-acetilglutamato que es un activador de la carbamil fosfato sintetasa I.
10. Defectos genéticos en el ciclo de la Urea:
Deficiencia o alteración en alguna enzima del ciclo de la urea = intolerancia a la ingesta de determinados
aminoácidos => acumulación de intermediarios del ciclo = hiperamonemia, la eliminación de las proteínas de
la dieta no es viable.
Deficiencia de ornitina transcarbamoilasa: Acumulacion de amoniaco y aminoácidos = retraso
mental y muerte, tratamiento suplementando la dieta con arginina y suplementos de aa esenciales.
Deficiencia de arginina succinato sintetasa: Aumento de la citrulina en sangre => citrulinemia,
tratamiento con suplementos de arginina.
Deficiencia de arginasa: Deficiencia en el SNC, tratamiento con aa esenciales.
Deficiencia de carbamoil-fosfato sintetasa: Hiperamonemia = retraso mental, tratamiento con
carbamil glutamato (análogo del activador).
Tratamiento de la hiperamonemia: Administracion de acidos aromáticos como el benzoato (a
benzoil-Coa + glicina = hipurato, la glicina debe regenerarse = gasto de amoniaco) o fenilbutirato
(pasa a fenilacetato por B-oxidacion = fenilacetil-CoA => combinación con glutamina = producción de
mas glutamina = gasto de amoniaco).
SEGUNDA PARTE: RUTAS DE DEGRADACION DE LOS AMINOACIDOS MIJAIL JN
15% de la producción energética, menor cantidad, formación de productos principales que convergen en el ciclo de
Krebs.
Aminoacidos glucogénicos: Cadena carbonada a piruvato o intermediarios de Krebs, pueden participar en
gluconeogénesis.
Aminoacidos cetogenicos: Cadena carbonada a acetil-coA que entra a Krebs o va como intermediario de síntesis
de acidos grasos.
COFACTORES
Transaminacion = Piridoxal fosfato.
Transferencia de grupos monocarbonados = Tetrahidrobiopterina (BH4), tetrahidrofolato(FH4), S-
adenosilmetionina (SAM).
Tetrahidrobipterina (BH4) => Transportador de e-.
Tetrahidrofolato (FH4) => Transferencia de grupos monocarbonados en oxidación intermedia, proviene del
folato por accion de la dihidrofolato reductasa, la forma reducida transporta metileno y la forma mas
oxidada transporta metileno, metenilo, formilo o forminino, Union al N5 o N10 = N5,N10-
metilenotetrahidrofolato.
11. S-adenosilmetionina (SAM o adoMet) => Transporte preferente de metilos, del ATP + metionina por accion
de metionina adenosil transferasa => Ataque nucleofilico del S de la metionina al C5 de la ribosa y liberación
de trifosfato.
Biotina: Transfiere carbono como CO2.
AMINOACIDOS QUE SE DEGRADAN A PIRUVATO
Alanina, Triptofano, Cisteina, Serina, Glicina, Treonina
Alanina y Triptofano:
Cisteina:
Serina:
Escisión de cadena lateral del
triptófano = Alanina
Transaminacion de alanina con α-cetoglutarato = Piruvato
1) Liberacion de azufre.
2)Transaminacion (Liberacion de NH3).
Serina Piruvato (accion de la serina
deshidratasa), reacción dependiende
de piridoxal fosfato.
12. Glicina:
Treonina:
ALTERACIONES EN EL CATABOLISMO:
Hiperglicinemia: Niveles elevados de glicina = Activacion de 2 receptores => Clasicos (medula
espinal, ocasiona apnea e hipotonía) y Corteza cerebral (ocasiona daño cerebral y convulsiones)
2 tipos: Cetosica y no cetosica
Cetosica: Bloqueo de la enzima de rotura de la glicina por inhibición externa (en
acidemias) => acidosis y cetosis.
1ra ruta:
Glicina -> Serina por
accion de serina
hidroximetil
transferasa (adicion
de un hidroximetilo)
2da ruta:
Ruptura oxidativa catalizada por enzima de rotura de la glicina
= CO2 + NH4 + CH2 (metileno) -> Acoplamiento del metileno al
tetrahidrofolato = N5,N10-Tetrahidrofolato
Coenzimas:
->Tetrahidrofolato
->Piridoxal fosfato
3ra ruta:
La glicina sustrato de la D-aminoacido oxidasa formado
glioxilato que se oxida a oxalato, esta via no se relaciona con
Krebs.
La treonina presenta 2 rutas:
Conversion a succinil-CoA (principal).
Conversion a piruvato via glicina.
Paso 1: Accion de la treonina
deshidrogenasa formado 2-amino-3-
cetobutirato o α-amino-β-cetobutirato
Paso 2: Accion de una ligasa
formado glicina.
Paso 3: Accion de la serina
hidroximetil transferasa formado
serina.
Paso 4: Accion de la serina
deshidratasa formado piruvato.
13. No cetosica: Es la mas común (1/250000), defectos congénitos (autosómico recesivo) en la
enzima de rotura de la glicina => concentraciones elevadas de glicina = deficiencias mentales
graves + muerte, contar que la glicina tmbn es un neurotransmisor inhibidor. (NKH > 0.09,
normal < 0.04).
Neonatal: Mas común, 6-8 dia, déficit en la succion + letargia + hipotonía profunda
+ convulsiones + mioclonias. A nivel cerebral: Hipogenesia, malformación en
circunvoluciones, crecimiento de ventrículos e hipoplasia. 30% Letal, secuelas =
retardo psicomotor y convulsiones.
Infantil: A partir del 6 mes de edad, inicialmente como convulsiones, forma mas
leve => mayor sobrevida.
Cristales de oxalato cálcico = 75% de los cálculos renales.
AMINOACIDOS QUE SE DEGRADAN A OXALACETATO
Asparagina y Aspartato
AMINOACIDOS QUE SE DEGRADAN A α-CETOGLUTARATO
Prolina, Glutamina, Glutamato, Arginina, Histidina
Paso 1: Accion de la asparaginasa sobre
la asparagina formando aspartato.
Paso 2: Transaminacion del Asparatato
con el α-cetoglutarato formando
glutamato y OXALACETATO.
El oxalacetato entra al ciclo de Krebs
oxidándose en CO2 + H2O generando ATP.
Dato: La asparaginasa es un
quimioterapéutico contra canceres que
necesitan asparagina = Leucemia
linfoblastica aguda.
14. AMINOACIDOS QUE SE DEGRADAN A SUCCINIL-CoA
Metionina, Isoleucina, Treonina, Valina
Primer paso,
introducción de
doble enlace en
la prolina por
acción de
oxidasa.
Segundo paso, de
estructura cíclica a
lineal por hidrolisis.
= Base de Schiff
Tercer paso, glutamato
semialdehido por accion de una
deshidrogenasa = Glutamato
Accion de la glutamato
deshidrogenasa =
DESAMINACION con el
glutamato formando α-
cetoglutarato.
Accion de la
glutaminasa con
la glutamina
formando
glutamato.Primer paso, Histidina Urocanato por
accion de histidina amoniaco liasa.
Segundo paso, Urocanato Imidazolona
propionato por accion de urocanato liasa.
Tercer paso, Imidazolona propionato N-
formiminoglutamato por accion de
imidazolana propionasa.
Cuarto paso, N-formiminoglutamato
Glutamato por accion de glutamato
fromiminotransferasa.
Primer paso, realizado dentro del
ciclo de la urea por accion de
arginasa formando Ornitina.
Segundo paso,
TRANSAMINACION de
la ornitina para
formar glutamato
semialdehido.
Tetrahidrofolato = Cofactor
15. Primer paso, Metionina + ATP con accion de
metionina adenosil transferasa = S-
adenosilmetionina (SAM/adoMet).
Segundo paso, SAM por accion de una metil
transferasa = S-adenosilhomocisteina.
Tercer paso, S-adenosilhomocisteina por
accion de hidrolasa produce Homocisteina.Cuarto paso, Homocisteina por accion de
cistationina sintasa incorpora una serina
formando CISTATIONINA.
Quinto paso, Cistationina por accion de una
liasa forma α-cetobutirato.
Treonina, Reaccion con la
treonina deshidratasa
para formar α-
cetobutirato.
La Isoleucina y la Valina empiezan con 3
reacciones con enzimas compartidas:
1) Transaminacion con α-cetoglutarato por
una Aminotransferasa.
2)Desaminacion oxidativa del producto por
una deshidrogenasa.
3)Accion de la acil-CoA deshidrogenasa
formando Tiglil-CoA y Metilacrilil-CoA.
Metilacrilil-CoA, 4 reacciones produciendo
Propionil-CoA.Tiglil-CoA, 3 reacciones
produciendo Propionil-CoA con la
liberación de un Acetil-CoA.
Carboxilacion del Propionil-CoA formando
Metilmalonil-CoA.
Accion de α-cetoacido deshidrogenasa sobre
α-cetobutirato formando Propionil-CoA.
Epimerizacion del Metilmalonil-CoA
formando Succinil-CoA por accion de
Metilmalonil-CoA mutasa (coenzima B12).
Las rutas degenerativas de Isoleucina y la
Valina relacionadas con pasos de la
degradación de ácidos grasos.
Homocisteina = Marcador de enfermedad,
hiperhomocisteinemia relacionada a
enfermedad cardiovasc + defectos del tubo
neural + anencefalia + espina bífida,
controlada con vit B6, B12 y folato.
16. AMINOACIDOS QUE SE DEGRADAN A ACETIL-CoA
Triptofano, Lisina, Fenilalanina, Tirosina, Leucina, Isoleucina, Treonina
La degeneración de Isoleucina produce
tanto Acetil-CoA como Propionil-CoA,
para más detalles ir a la pagina anterior.
Primer paso, Transaminacion de la leucina
formando acido α-cetoisocaproico.
Segundo paso, Desaminacion oxidativa del acido
α-cetoisocaproico formando isovaleril-CoA.
Tercer paso, Accion de acil-CoA deshidrogenasa
para formal β-metilcrotonil-CoA.
El β-metilcrotonil-CoA se degrada hacia Acetil-
CoA y Acetoacetato (que a la larga => Acetil-CoA)
Al introducir un CoA al acetoacetato
tenemos acetoacetil-CoA que si
introducimos otro CoA rompemos el
acetoacetil-CoA en 2 Acetil-CoA.
17. Primer paso, Conjugacion de la lisina con α-cetoglutarato (accion
de la sacaropina deshidrogenasa) formando SACAROPINA.
Segundo paso,
Deshidrogenacion de la
sacaropina formando
AMINOADIPATO
SEMIALDEHIDO +
glutamato.
Tercer paso, Oxidacion del aminoadipato
semialdehido formando α-AMINOADIPATO.
Cuarto paso,
Transaminacion con α-
cetoglutarato
formando α-
CETOADIPATO.
Quinto paso, Descarboxilacion
del α-cetoadipato formando
GLUTARIL-CoA.
Sexto paso, Deshidrogenacion
del glutaril-CoA formando un
doble enlace entre el C2=C3 =>
GLUTACONIL-CoA.
Septimo paso, Descarboxilacion del
glutaconil-CoA formando CROTONIL-CoA.
Octavo paso, Hidratacion del
crotonil-CoA formando β-
HIDROXIBUTIRIL-CoA.
Noveno paso, Deshidrogenacion del B-
hidroxibutiril-CoA formando ACETOACETIL-CoA.
Decimo paso, Introduccion de
un acetilo al C3 (Accion de la
HMG-CoA sintasa) del
acetoacetil-CoA formando
HMG-CoA.
Onceavo paso, Eliminacion
del ACETIL-CoA (en el C1 por
accion de la HMG-CoA liasa)
formando ACETOACETATO.
Una deficiencia en la sacaropina
deshidrogenasa lleva a
hiperlisinemia + hiperlisinuria =
Retraso mental
Primer paso, Accion de dioxigenasa formando N-
FORMILQUINURENINA.
Segundo paso, Accion de formidasa formando
QUINURENINA.
Tercer paso, Accion de monooxigenasa
formando 3-HIDROXIANTRANILATO.
Cuarto paso, Accion de quinureninasa (coenzima =
piridoxal pirofosfato) formando 3-HIDROXIQUINURENINA
+ ALANINA.
TRANSFORMACION de la 3-hidroxiquinurenina hacia α-
CETOADIPATO que continua la ruta a partir del QUINTO
PASO formando al final ACETOACETATO y ACETIL-CoA.
18. ALTERACIONES EN LA DEGRADACION DE LA FENILALANINA:
Defecto en la fenilalanina hidroxilasa => FENILCETONURIA (PKU) que causa hiperfenilalaninemia, en estos
casos entra en juego una ruta alternativa en la que la FENILALANINA se TRANSAMINA con PIRUVATO
formando ALANINA+FENILPIRUVATO, tanto la fenilalanina como el fenilpiruvato se acumulan en los tejidos
(ocasionando retraso mental grave, además de que compite con otros aminoácidos causando su déficit) y se
excretan por la orina, el fenilpiruvato se DESCARBOXILA a FENILACETATO que da un olor característico a esta
Primer paso, Reaccion de la fenilalanina
con la fenilalanina hidroxilasa (oxidasa
de función mixta) => hidroxilacion +
reducción simultanea, cofactor =
tetrahidrobiopterina que al actuar la
hidroxilasa este se reduce a
dihidrobipterina que por accion de una
reductasa con gasto de NADPH se
regenera a tetrahidrobiopterina.
Segundo paso, Transaminacion de
tirosina formando glutamato e
HIDROXIFENILPIRUVATO.
Tercer paso, Oxidacion del
hidroxifenilpiruvato formando
HOMOGENTISATO.
Cuarto paso, Oxidacion del
homogentisato formando
MALEILACETOACETATO.
Quinto paso, Isomerizacion del
maleilacetoacetato formando
FUMARILACETOACETATO.
Sexto paso, Ruptura del
fumarilacetoacetato formando
FUMARATO + ACETOACETATO.
19. orina; el tratamiento: Dieta con fenilalanina y tirosina únicamente suficientes para satisfacer necesidades +
restricción de alimentos ricos en proteínas + eliminación de la caseína + no consumir productos con
aspartame (dipeptido de aspartato + ester de fenilalanina).
Deficiencia de homogentisato deshidrogenasa => ALCAPTONURIA, el homogentisato no puede
metabolizarse llevando a su acumulación y provocando triada: Aciduria + Ocronosis (deposito de pigmento
marron-negruzco en el tejido conectivo) + Artritis prematura (puede llevar a anquilosis de la región
lumbosacra), se observa una orina café oscura + excreción de 7g/dia de acido homogentisico.
Defecto en tirosina 3-monooxigenasa => ALBINISMO, deficiencia en la síntesis de melanina a partir de la
tirosina llevando a falta de pigmentación = Cabello blanco y piel rosada.
DATO: Los intermediarios en degradación del triptófano son intermediarios para la síntesis de diversas biomoléculas.
20. TERCERA PARTE: BIOSINTESIS DE AMINOACIDOS MIJAIL JN
Recordar que los aminoácidos y por tanto el nitrógeno no se almacenan.
2 definiciones:
Dispensables verdaderos: Sintetizados a partir de otros aminoácidos o de metabolitos nitrogenados en
cantidades suficientes en cualquier circunstancia fisiológica o patológica.
Condicionalmente indispensables: Se pueden sintetizar de otros aminoácidos pero esta síntesis esta limitada
en ciertas circunstancias. Compuesto producido usualmente en cantidades adecuadas endógenamente pero
en ciertas circunstancias se requieren de forma exógena.
Esenciales: Deben necesariamente ser ingeridos ya que únicamente son producidos o bien por las plantas o
por las bacterias.
En el recién nacido:
AA no esenciales: Para síntesis de proteínas, forman intermediarios del ciclo de Krebs o de la urea y participan el la
gluconeogénesis. DATO: para producir cisteína se requieren grandes cantidades de metionina, lo mismo con la
fenilalanina para formar tirosina; HISTIDINA esencial en el RN.
Glutamato y glutamina punto de entrada, el glutamato fuente de aminos y el nitrógeno de la glutamina es fuente
de aminos para biosíntesis (transporte de amoniaco). Concentracion de ambos regulada en respuesta a necesidad y
para el equilibrio osmótico.
4 vias para biosintesis:
1. Glutamina sintetasa: NH4+glutamato = glutamina
Glutamato + NH4 + ATP => Glutamina + ADP + Pi + H
Es la via principal para incorporacion del NH4 al glutamato (papel central en el metabolismo de aminoácidos)
se encuentra en todos los organismos.
En el hígado y riñon Glutaminasa que rompe la glutamina en glutamato y amonio que entra al ciclo de la
urea para su excreción.
2. Glutamato sintasa: α-cetoglutarato aminacion reductora con glutamina formando 2 glutamatos, esta
enzima no esta presente en los animales.
21. 3. Transaminacion con α-cetoglutarato:
Piruvato Alanina; Oxalacetato Asparagina; Cetoglutarato Glutamato.
4. Glutamato deshidrogenasa:
La glutamato deshidrogenasa es reversible, se encuentra en la matriz mitocondrial. Para formar el glutamato
utiliza NADPH incorporando amonio al α-cetoglutarato. Activada por ATP y GTP en el sentido de síntesis de
glutamato.
Todos los aminoácidos proceden de intermediarios de glucolisis o del ciclo del acido citrico, el ser humano solo
puede sintetizar 10 de los 20 aminoacidos estándar => los que se sintetizan = aminoácidos no esenciales.
GLUTAMATO
DESHIDROGENASA
NADH
NH4H2ONAD
22. SINTESIS DE ALANINA, ASPARAGINA, ASPARTATO, GLUTAMATO Y GLUTAMINA
Precursores: Piruvato=>Alanina; Oxalacetato=>Aspartato=>Asparagina; a-cetoglutarato=>Glutamato =>Glutamina.
SINTESIS DE PROLINA, ORNITINA Y ARGININA
Precursor: Glutamato.
La ALANINA se sintetiza a partir de
PIRUVATO mediante una reacción de
transaminacion.
El ASPARTATO se sintetiza a partir del
OXALACETATO mediante una reacción
de transaminacion.
La ASPARAGINA se sintetiza mediante
la aminacion (amino de la glutamina)
del ASPARTATO por accion de
ASPARAGINA SINTETASA.
Ya vimos la síntesis del GLUTAMATO,
este por accion de la GLUTAMINA
SINTASA forma GLUTAMINA.
Dato: Los linfocitos malignos en
leucemia linfoblastica aguda infantil
requieren asparagina para su
crecimiento, bloqueando la síntesis
de asparagina junto a quimioterapia
= remisión del 95% casos, pero
pueden haber recaidas.
Primer paso, fosforilacion del
carboxilo del glutamato por accion de
una cinasa = Glutamato-5-fosfato.
Segundo paso, deshidrogenacion
(reducción) del glutamato-5-fosfato =
Glutamato-5-semialdehido.
Primer paso, ciclación espontanea del
glutamato-5-semialdehido = delta 1-
pirrolina-5-carboxilato (P5C) esta es
una base de Schiff.
Segundo paso, reducción del
P5C = Prolina.
TRANSAMINACION del Glutamato-
5-semialdehido con glutamato =
Ornitina.
ENTRADA AL CICLO DE LA UREA y
producción de Ornitina.
23. SINTESIS DE SERINA, CISTEINA Y GLICINA
Precursor: 3-Fosfoglicerato.
La via que acabamos de describir para la síntesis de cisteína se llama también: VIA DE TRANSULFURACION, la
CISTEINA producida se usa para sintetizar proteínas y glutatión, aumento serico de cisteína = ateroesclerosis.
SINTESIS DE TIROSINA A PARTIR DE FENILALANINA
Primer paso, oxidación del hidroxilo del 3 fosfoglicerato
por una deshidrogenasa = 3-Fosfohidroxipiruvato
Segundo paso, transaminacion con
glutamato = 3-Fosfoserina.
Tercer paso, hidrolisis de la 3-
fosfoserina para liberar el fosfato =
Serina.
La SERINA se convierte a GLICINA al
eliminar un carbono por la serina
hidroximetiltransferasa que tiene
como cofactor al tetrahidrofolato
(acepta el carbono saliente), es
reversible y requiere PLP (Fosfato de
piridoxal).
La CISTEINA = METIONA (da el S) +
SERINA (esqueleto carbonado).
PASO PREVIO
1. Metionina -> S-adenosilmetionina
(SAM o adoMet).
2. S-adenosilmetionina cede su metilo
= S-adenosilhomocisteina (adoHcy).
3. AdoHcy se hidroliza = homocisteina.
Paso 1: Homocisteina + Serina =
Cistationina por accion de cistationina
sintasa.
Paso 2: Cistationina liasa reacciona
con la cistationina (coenzima PLP) =
CISTEINA + α-cetobutirato.
24. Son vias complejas de síntesis pero en los animales se produce la tirosina directamente de fenilalanina por
hidroxilacion del fenilo en C4 por accion de FENILALANINA HIDROXILASA (coenzima = tetrahidrobiopterina).
Recordar que la tirosina = condicionalmente esencial y fenilalanina = esencial.
VIAS METABOLICAS DE LOS AMINOACIDOS ALIFATICOS, AROMATICOS Y RAMIFICADOS
FAMILIA DEL GLUTAMATO
Procedente de intermediario de Krebs: α-cetoglutarato que se convierte a glutamato por desaminacion
oxidativa o por transaminacion.
Alanina aminotransferasa (ALT) = Glutamato – piruvato transaminasa (GPT), específicamente
hepática, menor en hepatitis vírica pero mayor en cirrosis, enfermedad hepática, tumores hepáticos,
colestasis y hepatitis toxica.
glutamato + piruvato ⇌ α-cetoglutarato + alanina
Glutamato deshidrogenasa:
RECORDAR QUE PARTICIPA EN EL CICLO DE LA UREA.
Del glutamato se obtiene la glutamina que puede pasar a formar triptófano, además da el N3 y N9 para
formar los nucleótidos => PURINAS.
GLUTAMATO
DESHIDROGENASA
NADH
NH4H2ONAD
25. FAMILIA DEL OXALACETATO/ASPARTATO
El oxalacetato forma el aspartato mediante transaminacion:
Aspartato aminotransferasa (AST) = Glutamato – oxalacetato transaminasa (GOT) en corazón,
hígado y musculo; en cantidades elevadas en infarto agudo de miocardio, hepatopatía y miopatías
(daño celular grave).
L-aspartato + 2-oxoglutarato ⇌ oxalacetato + L-glutamato
La treonina deriva en Isoleucina.
FAMILIA DEL 3-FOSFOGLICERATO/SERINA
El 3-fosfoglicerato es un intermediario de la glucolisis.
Recordar que la serina también participa como grupo cabeza en la síntesis de Fosfatidiletanolamina a
partir de Fosfatidilserina (que únicamente se descarboxila).
FAMILIA DEL PIRUVATO
Los aminoácidos reaccionan con el piruvato mediante transaminacion.
Alanina aminotransferasa (ALT) = Glutamato – piruvato transaminasa (GPT), específicamente
hepática, menor en hepatitis vírica pero mayor en cirrosis, enfermedad hepática, tumores hepáticos,
colestasis y hepatitis toxica.
glutamato + piruvato ⇌ α-cetoglutarato + alanina
Intermediario en la síntesis de la Valina es el α-cetoisovalerato el que es punto de partida de la via
colateral que lleva a la leucina.
26. FAMILIA DE LOS AROMATICOS
El fosfoenolpiruvato proviene de la glucolisis y la Eritrosa 4-fosfato proviene de la via de las pentosas
fosfato.
FAMILIA DE LA HISTIDINA
CUARTA PARTE: MOLECULAS DERIVADAS DE AMINOACIDOS MIJAIL JN
Aminoacidos = bloques de construcción + precursores de biomoléculas (hormonas, coenzimas, nucleótidos,
porfirinas, neurotransmisores, etc).
NEUROTRANSMISORES DERIVADOS DE LA TIROSINA
La tirosina origina a catecolaminas = DOPAMINA, NORADRENALINA (noreprinefrina), ADRENALINA (epinefrina).
Paso 1: Formacion de DOPA por accion
de una hidroxilasa (coenzima =
tetrahidrobiopterina) que introduce un
hidroxilo en el anillo.
Paso 2: Formacion de DOPAMINA por
accion de una descarboxilasa
(coenzima = piridoxal fosfato o PLP)
que introduce un hidroxilo en el anillo.
La dopamina se encuentra en el nucleo
caudado, putamen y globo palido (SNC),
disminuido en Parkinson y aumentado en
Esquizofrenia.
La dopamina no difunde por la barrera
hematoencefalica asi que para el Parkinson se
administra DOPA.
Paso 3: Formacion de NORADRENALINA
por accion de una hidroxilasa, para esta
reacción se requiere O2 + Ascorbato
(vit.C) y Cu.
La noradrenalina es el principal
neurotransmisor simpático
posganglionar, actua sobre los
receptores alfa y tiene efecto
vasopresor.
Paso 4: Formacion de ADRENALINA por
transmetilacion, se requiere adoMet o
SAM como dador de metilo. LA ENZIMA
ESTA EN LA MEDULA ADRENAL.
La adrenalina NO es mediador en la
neurona simpática posganglionar,
tiene accion sobre receptores alfa y
beta + aumento de frecuencia
cardiaca, estimula glucogenolisis y
lipolisis.
27. Vida media de 15 a 30 segundos, inactivadas por la monoamino oxidasa (MAO) y catecol-O-metil
transferasa (COMT), la MAO es la única que se encuentra en las terminales nerviosas.
SINTESIS DE MELANINA
La melanina da el color a la piel y al pelo, se sintetiza de la siguiente manera: tras la hidroxilacion de la tirosina a
DOPA (que en los melanocitos catalizada por tirosinasa) que tras varias oxidaciones no enzimáticas se polimeriza en
pigmentos de color marron y negro.
Tipos de melanina:
Eumelanina: Piel oscura, iris y cabello.
Feomelaninas: Piel, iris y cabello rojo, labio, pezones y genitales.
Neuromelaninas: Neuronas.
Tricocromos: Pelo y plumas.
SINTESIS DEL GABA
Neurotransmisor inhibitorio, mayor nivel en el cerebelo y poco nivel en el talamo + hipocampo. Alteraciones
relacionadas a corea de Huntington, Parkinson, Alzheimer, demencia senil y esquizofrenia.
Producido por la descarboxilacion del glutamato.
Dato: Deficiencia => Epilepsia.
SINTESIS DE LA SEROTONINA
Formada del triptófano en el SNC, plaquetas y tracto gastrointestinal.
28. SINTESIS DE LA HISTAMINA
Amina idazolica de la descarboxilacion de la histidina, potente vasodilatador liberado en respuesta alérgica, estimula
también la secreción acida del estomago.
POLIAMINAS
Putrescina o 1,4 diaminobutano, al descomponerse la carne junto a la cadaverina formadas por la descomposición
de aminoácidos, producida en pequeñas cantidades por las células vivas por accion de ornitina descarboxilasa
funcionando como factor de crecimiento.
Espermina y espermidina, derivadas de la metionina y ornitina, tienen un papel en el empaquetamiento del ADN.
El primer paso es la descarboxilacion de la ornitina formando putrescina que por accion de propilaminotransferasa I
forma espermidina que por accion de la propilaminotransferasa II forma la espermina.
FORMACION DE LA FOSFOCREATINA
Deriva de la creatina, amortiguador energético en el musculo (puede donar un fosfato al ADP = ATP mediado por
creatin quinasa), esta creatina deriva de la glicina y arginina.
GLICINA + ARGININA => accion de amidinotransferasa => GUANIDINOACETATO => accion de metiltransferasa (junto
a adoMet o SAM) => CREATINA => accion de creatina quinasa => FOSFOCREATINA
La creatina se excreta via renal, medición para determinar la función renal.
29. NEUROTRANSMISORES DERIVADOS DEL TRIPTOFANO
Triptofano => Serotonina (en las plaquetas y tracto gastrointestinal en mayor cantidad, tmbn en el cerebro y retina)
+ Melatonina (derivado de la serotonina en la hipófisis y retina => N-acetiltransferasa, aumenta durante la noche =
variación diurna que es manejada por la norepinefrina).
30. QUINTA PARTE: PORFIRINAS Y METABOLISMO DE LA BILIRRUBINA MIJAIL JN
Las porfirinas tienen un precursor = Glicina, estas porfirinas contribuyen con la formación de la Hb y citocromos.
En las plantas y algas el precursor es el glutamato.
La glicina reacciona con el Succinil-CoA mediante la delta-aminolevulinato (5-aminolevulinato) sintasa formando un
intermediario = α-amino-β-cetoadipato que se descarboxila por la misma enzima para formar DELTA-
AMINOLEVULINATO (5-AMINOLEVULINATO o ALA) que pasa al citosol para iniciar la síntesis.
PRIMER PAS CITOSOLICO: Union de 2 moleculas de 5-aminolevulinato (ALA) formando porfobilinogeno por
accion de porfobilinogeno sintasa.
SEGUNDO PASO CITOSOLICO: Union de 4 porfobilinogenos por accion de hidroximetilbilano sintasa liberando
amonio y formando en primera instancia el Hidroximetilbilano (estructura tetrapirrolica lineal) que cicla
espontáneamente en Uroporfirinogeno I o puede pasar a Uroporfirinogeno III (únicamente este isómero
continua la síntesis) por accion de una sintasa.
TERCER PASO CITOSOLICO: El uroporfirinogeno III por accion de una descarboxilasa que convierte el acido
acético a metilo se transforma en Coproporfirinogeno III (Copro III), esto sucede análogamente con el Uro I
que se transforma a Copro I.
31. PRIMER PASO MITOCONDRIAL: Descarboxilacion (enzima en la membrana interna de la mitoc.) del
coproporfirinogeno III a nivel de 2 de los acidos propionicos convirtiéndolos en vinilo formando
Protoporfirinogeno IX.
SEGUNDO PASO MITOCONDRIAL: Oxidacion = inserción de dobles enlaces en el Protoporgirinogeno IX por
accion de una oxidasa => Protoporfirina IX.
CUARTO PASO MITOCONDRIAL: La protoporfirina IX reacciona con la ferroquelatasa que introduce el Hierro
ferroso (Fe2+) en el anillo formando el Hemo.
REGULACION DE BIOSINTESIS DEL HEM
En el hepatocito el paso limitante = reaaccion de ALA sintasa, el hemo es correpresor junto a la hemina (derivada de
oxidación del hierro férrico)
En los globulos rojos ocurre a varios niveles: ALA sintasa (menos hierro ferroso inhibe, mas hierro ferroso activa) y en
la ferroquelatasa.
32. PORFIRIAS
Defectos enzimáticos que llevan a acumulación de precursores en el eritrocito, fluidos corporales e hígado.
TIPOS
NEUROPSIQUIATRICAS CUTANEAS MIXTAS
Porfiria aguda intermitente
(PAI)
Porfiria cutánea tardia
(PCT)
Coproporfiria hereditaria
(CPH)
Plumboporfiria Porfiria eritropoyetica
congénita
(PEC)
Porfiria variegata o jaspeada
(PV)
Protoporfiria eritropoyetica
(PPE)
Porfiria hepatoeritropoyetica
(PHE)
Sin síntomas fotocutaneos: Acumulacion de precursores de porfirinas => Porfiria de Doss o plumboporfiria y
la PAI.
Con síntomas fotocutaneos: Acumulacion de porfirinas => Las restantes.
AGUDAS: Plumboporfiria (deficiencia en ALA-deshidratasa), PAI, CPH y PV.
NO AGUDAS: Las restantes.
PORFIRIAS AGUDAS
Deficiencia en la ALA-deshidratasa o Plumboporfiria o de Doss: Es la mas rara (solo 7 casos), autosómica
recesiva, muy similar a la PAI, aumento en excreción de ALA por la orina.
PAI (Porfiria Aguda Intermitente): Mas frecuente, autosómica dominante por deficiencia en la PBG
desaminasa, mas en mujeres tras la pubertad, NO PRESENTA SINTOMAS CUTANEOS, se observa
hiponatremia con orina rojiza, nauseas, debilidad muscular. Sintomas desencadenados por menor ingesta
calórica, drogas, alcohol, tabaco, infecciones, estrés y hormonas. Excrecion de ALA, PBG Y PBGD
aumentadas.
CPH (Coproporfiria Hereditaria): Muy rara, autosómica dominante, deficiencia en la coproporfirinogeno
oxidasa, mas en mujeres tras la pubertad, síntomas similares a la PAI pero con fotosensibilidad,
Coproporfirina III eliminada en heces.
PV (Porfiria Variegata o jaspeada): Autosomica dominante con mayor predominio en sudafrica, deficiencia
en la protoporfirinogeno oxidasa, se observan síntomas neuropsiquiatricos y cutáneos, aumento de
porfirinas biliares aumentando el riesgo de litiasis.
PORFIRIAS NO AGUDAS:
PEC (Porfiria Eritropoyetica Congenita): Deficiencia en uroporfirinogeno III sintetasa, muy rara, autosómica
dominante con severas manifestaciones cutáneas, anemia hemolítica y esplenomegalia, mas coproporfirina I
y uroporfirina, ALA y PBG normales.
33. PCT (Porfiria Cutanea Tarda): Deficiencia en uroporfirinogeno descarboxilasa, mas frecuente, autosómica
dominante, Adquirida (Tipo I)/Hereditaria (Tipo II) = 4/1. Desencadenada por drogas, alcohol, estrógenos,
VIH, hierro y hemocromatosis. Mas uroporfirina I e Isocoproporfirina, ALA y PBG normales, pueden
desarrollar cirrosis y cáncer hepático.
PHE (Porfiria Hepatoeritropoyetica): Deficiencia en uroporfirinogeno descarboxilasa, muy rara, variante
recesiva de PCT tipo II, en la niñez con severa fotosensibilidad junto a anemia hemolítica y esplenomegalia.
Aumento de uroporfirina e isocoproporfirina, ALA y PBG normales.
PPE (Protoporfiria eritropoyetica): Deficiencia en la ferroquelatasa, autosómica dominante con síntomas
cutáneos desde la niñez sin síntomas neurológicos, aumento de protoporfirina sérica y en heces mientras
que en la orina normal, puede presentarse colestasia y daño hepático.
METABOLISMO DEL HEMO
Bilirrubina => De degradación del hemo liberado tras la lisis de eritrocitos, 80% de la Hb (250-400mg) y el 20% de
otras hemoproteinas (mioglobina, hemocianina, clorocruorina, peroxidasa, citocromos, citocromo c).
NIVELES:
TOTAL = 0.3 a 1.1 mg/dl
DIRECTA = 0.1 a 0.4 mg/dl
INDIRECTA = 0.2 a 0.7 mg/dl
Paso 1: Accion de la hemo oxigenasa
convierte el Hemo (cíclico) a
Biliverdina (lineal) liberando CO y
Fe2+.
Paso 2: Accion de la biliverdina
reductasa deshidrogena la biliverdina
a Bilirrubina libre o Indirecta.
Paso 3: La bilirrubina indirecta es muy
insoluble y viaja por via sanguínea
unida con la albumina (seroalbumina)
hasta el hígado.
Paso 4: Conjugacion (accion de
glucoronil bilirrubina transferasa) con
acido glucuronico (80%) o acido
sulfúrico (20%) formando Bilirrubina
directa que es soluble.
Paso 5: La bilirrubina sale junto a las
demás sales biliares al intestino.
5% REABSORBIDO , llega al riñon y se
excreta como urobilina. Paso 6: Enzimas bacterianas
Urobilinogeno=>Estercobilinogeno
=>Estercobilina
34. ICTERICIA
Pigmentacion amarillenta de piel y mucosas causa por exceso de acumulación de bilirrubina.
DATO: Falsa ictericia => Ingestion de alimentos ricos en carotenos o licopenos.
PRE-HEPATICA
Aumento de
Bilirrubina indirecta
Sobreproduccion
Anemia hemolítica
(autoinmune o
esferocitosis).
Talasemias.
Anemia
megaloblastica.
Absorcion de
hematomas.
La bilirrubina
indirecta es
insoluble por tanto
esta unida a la Hb y
no filtra.
HEPATICA
Aumento de
Bilirrubina indirecta
Menor captación Sindrome de Gilbert
Aumento en la
excreción de la
bilirrubina directa
ya que esta es
soluble y es filtrada
por el riñon.
Menor conjugación
Enfermedad de
Crigler Najar
Aumento de la
Bilirrubina directa
Alteracion en
transporte y
excreción
Sindrome de Dubin-
Johnson
Sindrome Rotor
Obstruccion en via
biliar intrahepatica
Cirrosis Biliar
Necrosis
hepatocelular
Hepatitis
POST-HEPATICA
Aumento de la
Bilirrubina directa
Obstruccion en via
biliar extrahepatica
Calculos biliares
Calculos de cabeza
de pancreas
35.
36. SEXTA PARTE: NUTRICION MIJAIL JN
ROL DE LOS AMINOACIDOS
ARGININA: Sintetizada en el organismo pero su deficiencia => menor crecimiento, soluciones para
nutrición parental sin este aminoácido => hiperamonemia, acidosis metabolica y coma, síntesis disminuida
en RN prematuros.
Requerimientos: 4.4 a 10.1 g/dia
Ingesta suboptima: <2.6 g/dia
En: pescado, carne, concentrados de arroz y leguminosas, frutos secos, sandia y algas. Leche poca arginina.
Funciones: Sintesis (de proteínas, creatina, NO, agmantina), regulación hormonal (insulina, glucagón, GH,
etc), regulación (de inmunidad, neuronal, reproducción, metabolismo energentico, tisular).
Concentracion en hígado baja: 0.03 a 0.1 mmol.
La arginina puede provenir de GLUTAMINA, GLUTAMATO O PROLINA séricas que pasan a los enterocitos y se
degradan a citrulina que llega al riñon donde se transforman en ARGININA que vuelve a la sangre (el 40% no
lo hace).
Destinos metabólicos:
Sintesis proteica: demanda>disponibilidad => Crecimiento acelerado (RN) o estrés metabolico
(prolif. Celular).
Ciclo de la urea: Regulacion + en ureogenesis, deficiencia afecta producción de urea.
Oxido nítrico: Sustrato de la NO sintasa, el NO es vasodilatador, antiaterogenico y antiagregante.
Creatinina: Almacen energético, consume 2.3 g/dia de arginina para su síntesis.
Aginina + Glicina => Ornitina + Acido guanidoacetico, el Acido guanidoacetico reacciona con el
adoMet o SAM para formar creatina.
Otros proceso: Extrahepaticamente por Arginasa II para la regulación de síntesis de NO, prolina
(precursor de la síntesis del colágeno por tanto en quemados o diabéticos la administración de
arginina => curación), poliaminas (activan proliferación celular).
CISTEINA: AA dispensable, sintetizado de metionina y serina, en el RN prematuro es condicionalmente
esencial (menos actividad de cistationasa hepática) por ello si hay algún problema se les alimenta con leche
formula enriquecida con cisteína. Administracion exógena para formación de glutatión, en enfermedades
hepáticas baja actividad de cistationasa => administración de cisteína, en su síntesis produce SH2 que es un
señalizador celular.
GLICINA: AA glucogénico precursor de creatina, profirinas, glutatión y nucleótidos. Abundante en el
colágeno, regula entrada de Ca.
Requerimientos: 3.2g/dia
Condicionalmente esencial en RN prematuros, requerimientos de este pueden no ser satisfechos por la leche
materna.
37. GLUTAMINA: AA no esencial pero en estrés metabolico (sepsis, estrés quirúrgico o politraumatismo)
aumento de demanda de glutamina, + abundante en sangre (650mmol/L), en mayor cantidad en cels (61%
de prot muscular), ½ de aa corporales, transporte de N.
Funciones: Regula secreción de insulina, menos glucocorticoides, síntesis de nucleótidos (da el N3 y
N9), reservorio de N (detoxificacion del amoniaco), inhibe la apoptosis, síntesis de aminoazucares y
equilibrio acido-base (la glutamina => iones amonio => eq acido-basico).
De la glutamina deriva el GABA, folatos y el glutamato.
PROLINA: AA no esencial, del glutamato y visceversa, puede hidroxilarse para contribuir a formación de
colágeno, papel en el sistema inmunitario intestinal (prolina oxidada => H2O2 citotoxico para bacterias).
Requerimientos: 5.2g/dia, jóvenes pueden ingerir hasta 12g/dia.
TAURINA: Ac. B-aminoetanosulfonico, no proteinogenico, amina intracel + abundante, 70kg peso = 70g de
taurina a 5-50mmol de concentración intracelular y 100 umol plasmático.
Funciones: Formacion de acidos biliares (por tanto excreción de colesterol), osmorregulacion, inhibe
fosforilacion de proteínas, función retiniana, desarrollo fetal y del RN.
Metabolismo principalmente en hígado.
Extrahepaticamente de N-acetilcisteina y glutatión.
Requerimientos: menos de 200mg/dia.
TIROSINA: AA no esencial pero en enfermedad hepática o insuficiencia renal o en el RN(condicionalmente
esencial), modula la respuesta inmunitaria => Produccion de catecolaminas + tiroxina.
Requerimientos: 2.8g/dia hasta 6.4g/dia.
DERIVADOS DE AA CONDICIONALMENTE ESENCIALES
CARNITINA: Deriva de lisina y metionina, mas en el tejido muscular, en la B-oxidacion (lanzadera a
la matriz mitocondrial), deficiencia => Debilidad muscular + infiltración lipídica + menos carnitina
muscular => Cardiomiopatia, hipoglucemia, hiperamonemia, menor cetogenesis.
Fuentes: Carnes rojas, leche y pescado. Casi nada en vegetales.
Deficiencia: Sistemica (raro) o Miopatica (solo al tejido muscular).
Causas: Deficiencia en Lisina o Metionina, deficiencia en Fe o Vit C o B3/B6, fallo genético,
aciduria o problemas renales.
Suplementos con carnitina mejora complicaciones cardiacas y capacidad para ejercicio.
En el RN es condicionalmente esencial => Suplementacion.
COLINA: Amina cuaternaria (trimetiletanolamina), parte de fosfolípidos, para neurotransmisión y
señalización celular.
Fuentes: Carnes rojas, vísceras, huevo, frutos secos y vegetales.
Requerimientos:
Lactantes 125-150mg/dia
Niños 200-375mg/dia
Varones 550mg/dia
Mujeres 400-425mg/dia
Gestacion 450mg/dia
Lactancia 550mg/dia
B-HIDROXI B-METILBUTIRATO: Metabolito de leucina, producción endógena en el citosol
muscular y hepático, contribuye a síntesis de colesterol. Su suplemento con la dieta disminuye el
daño muscular.
POLIAMINAS: Derivan de ornitina y metionina, para multiplicación y crecimiento celular =>
Putrescina, espermidina y espermina. Estabilizan ADN y ARN => empaquetamiento.
38. REQUERIMIENTOS PROTEICOS
Multiples funciones entre ellas gluconeogénesis (via alanina), forman oxidasas, para formación de anticuerpos y
forman homonas, neurotransmisores y proteínas plasmáticas.
REQUERIMIENTO = 0.8 g/Kg/dia
Si: 70Kg => 0.8x70 = 56g/dia
70% animal (39.2g) y 30% vegetal (16.8g)
En el embarazo (+10g/dia) y en la lactancia => Primeros 6 meses +15g/dia y segundos 6 meses +12g/dia.
En hipoproteinemia 1-1.2g/kg/dia.
En estrés 2g/kg/dia.
AA ESENCIALES => ADULTO = 84 mg/Kg/dia
AA ESENCIALES => LACTANTE = 714 mg/Kg/dia
PROTEINAS COMPLETAS
Aseguran crecimiento + reproducción + supervivencia + salud + % optimo de aa
Algunas deficientes en Lisina, triptófano, metionina o treonina.
Queso, huevos, leche y carne presentan todas excepto metionina, fenilalanina y triptófano.
Cereales presentan metionina.
Triptofano y metionina en semillas de ajonjolí y girasol.
Debe haber combinación para resolver las deficiencias. El glutámico = Arginina, Cistina = Metionina, Tirosina = ½
fenilalanina.
FUENTES DE PROTEINAS
LACTEOS: Mas el queso (hasta 39.1g%), menos la leche (3.1g%).
CARNES: Mas la carne de vacuno (25g%)y menos ciertas vísceras (11-17g%) + clara de huevo (11%).
VERDURAS: Mas el mani (26g% fresco, menos 10g% tostado) y menos las legumbres (menos de 2g%). El
tarhui y la soya 29% de proteínas.
CEREALES: De 7 a 14g%, Trigo no lisina, Maiz no triptófano y Arroz no Triptofano, cistina, metionina. La
quinua 11% de proteínas.
Papa y platano solo 2% proteínas, la yuca 1% de proteínas y la leche 3% de proteínas.
VALOR BIOLOGICO = (NITROGENO RETENIDO/NITROGENO ABSORBIDO) X 100
Las proteínas de origen animal VB + alto (Huevo 100%, Leche 85% y Carnes 75%).
Las proteínas de origen vegetal VB + bajo (Cereales 65%, Leguminosas 60%, Hortalizas 55%).
BN= NITROGENO INGERIDO – [NITROGENO UREICO + NITROGENO CREATININICO + NITROGENO FECAL (2g)]
Tipos:
BN 0 => Ingesta = Excreta
BN (-) => Ingesta < Excreta => En inanición, desnutrición, vejez, diabetes no controlada, fiebre, estrés (sepsis,
post quirúrgico, traumatismo, quemaduras).
BN (+) => Ingesta > Excreta => En niñez (crecimiento), gestantes, pacientes en recuperación y post inanición.
Valores:
>2 = Anabolismo
2 a -2 = Equilibrio
-2 a -5 = Catabolismo leve
-5 a -10 = Catabolismo moderado
< -10 = Catabolismo severo
KWASHIORKOR
En niños de 1-3 años, dieta baja en proteínas pero ingesta de CH normal o excesiva.
Sintomatologia: Irritabilidad, diarrea, falta de crecimiento, infecciones, cambios en el color del pelo, piel escamosa,
infiltración grasa en el hígado, menor masa muscular y edema masivo.
MARASMO
Carencia de alimentos en general (poca proteínas y poco CH), mas común en el primer año de edad.
Cuasado también por infecciones o parasitosis, también parto prematuro, malabsorción o interrupción temprana de
la lactancia.
39. Sintomatologia: Crecimiento deficiente, poca grasa subcutánea, delgadez extrema, anemia, ulceraciones por presión,
cambio en la textura del cabello el color normal.
KWASHIORKOR MARASMICO
Malnutricion grave con edema y peso menor al 60% de lo esperado de su edad. Presentan caracterisiticas del
marasmo y edema, además se puede observar dermatosis y hepatomegalia.
Característica Kwashiorkor Marasmo
Insuficiente crecimiento Presente Presente
Emaciación Presente Presente, notorio
Edema Presente (algunas veces leve) Ausente
Cambios en el cabello Común Menos común
Cambios mentales Muy común Raros
Dermatosis, copos de pintura Común No ocurre
Apetito Pobre Bueno
Anemia Grave (algunas veces) Presente, menos grave
Grasa subcutánea Reducida pero presente Ausente
Rostro Puede ser edematoso Macilento, cara de mono
Infiltración grasa del hígado Presente Ausente