Hidden Markov Models Explained
•Als PPT, PDF herunterladen•
1 gefällt mir•958 views

Hidden Markov models can be used to model sequential data and detect patterns. The document describes an HMM to detect CpG islands in DNA sequences. It has two states, "CpG island" and "not CpG island". Transition and emission probabilities are estimated from training data. The Viterbi, forward-backward, and Baum-Welch algorithms are used to find the most likely state sequence and re-estimate parameters when the true state sequence is unknown. The model can be extended to higher-order HMMs and different state duration distributions.

Empfohlen
Empfohlen
Weitere ähnliche Inhalte
Was ist angesagt?
Was ist angesagt? (20)
Ähnlich wie Hidden Markov Models Explained
Ähnlich wie Hidden Markov Models Explained (20)
Mehr von BioinformaticsInstitute
Mehr von BioinformaticsInstitute (20)
Kürzlich hochgeladen
Kürzlich hochgeladen (20)
Hidden Markov Models Explained
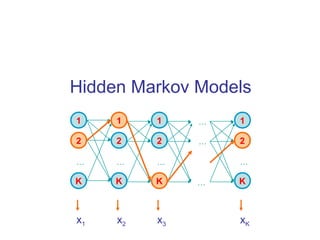
- 1. Hidden Markov Models 1 2 K … 1 2 K … 1 2 K … … … … 1 2 K … x1 x2 x3 xK 2 1 K 2
- 2. CS262 Lecture 6, Win06, Batzoglou Viterbi, Forward, Backward VITERBI Initialization: V0(0) = 1 Vk(0) = 0, for all k > 0 Iteration: Vl(i) = el(xi) maxk Vk(i-1) akl Termination: P(x, π*) = maxk Vk(N) FORWARD Initialization: f0(0) = 1 fk(0) = 0, for all k > 0 Iteration: fl(i) = el(xi) Σk fk(i-1) akl Termination: P(x) = Σk fk(N) ak0 BACKWARD Initialization: bk(N) = ak0, for all k Iteration: bl(i) = Σk el(xi+1) akl bk(i+1) Termination: P(x) = Σk a0k ek(x1) bk(1)
- 3. CS262 Lecture 6, Win06, Batzoglou A+ C+ G+ T+ A- C- G- T- A modeling Example CpG islands in DNA sequences
- 4. CS262 Lecture 6, Win06, Batzoglou Methylation & Silencing • One way cells differentiate is methylation Addition of CH3in C-nucleotides Silences genes in region • CG (denoted CpG) often mutates to TG, when methylated • In each cell, one copy of X is silenced, methylation plays role • Methylation is inherited during cell division
- 5. CS262 Lecture 6, Win06, Batzoglou Example: CpG Islands CpG nucleotides in the genome are frequently methylated (Write CpG not to confuse with CG base pair) C → methyl-C → T Methylation often suppressed around genes, promoters → CpG islands
- 6. CS262 Lecture 6, Win06, Batzoglou Example: CpG Islands • In CpG islands, CG is more frequent Other pairs (AA, AG, AT…) have different frequencies Question: Detect CpG islands computationally
- 7. CS262 Lecture 6, Win06, Batzoglou A model of CpG Islands – (1) Architecture A+ C+ G+ T+ A- C- G- T- CpG Island Not CpG Island
- 8. CS262 Lecture 6, Win06, Batzoglou A model of CpG Islands – (2) Transitions How do we estimate parameters of the model? Emission probabilities: 1/0 1. Transition probabilities within CpG islands Established from known CpG islands (Training Set) 2. Transition probabilities within other regions Established from known non-CpG islands (Training Set) Note: these transitions out of each state add up to one—no room for transitions between (+) and (-) states + A C G T A .180 .274 .426 .120 C .171 .368 .274 .188 G .161 .339 .375 .125 T .079 .355 .384 .182 - A C G T A .300 .205 .285 .210 C .233 .298 .078 .302 G .248 .246 .298 .208 T .177 .239 .292 .292 = 1 = 1 = 1 = 1 = 1 = 1 = 1 = 1
- 9. CS262 Lecture 6, Win06, Batzoglou Log Likehoods— Telling “Prediction” from “Random” A C G T A -0.740 +0.419 +0.580 -0.803 C -0.913 +0.302 +1.812 -0.685 G -0.624 +0.461 +0.331 -0.730 T -1.169 +0.573 +0.393 -0.679 Another way to see effects of transitions: Log likelihoods L(u, v) = log[ P(uv | + ) / P(uv | -) ] Given a region x = x1…xN A quick-&-dirty way to decide whether entire x is CpG P(x is CpG) > P(x is not CpG) ⇒ Σi L(xi, xi+1) > 0
- 10. CS262 Lecture 6, Win06, Batzoglou A model of CpG Islands – (2) Transitions • What about transitions between (+) and (-) states? • They affect Avg. length of CpG island Avg. separation between two CpG islands X Y 1-p 1-q p q Length distribution of region X: P[lX = 1] = 1-p P[lX = 2] = p(1-p) … P[lX= k] = pk-1 (1-p) E[lX] = 1/(1-p) Geometric distribution, with mean 1/ (1-p)
- 11. CS262 Lecture 6, Win06, Batzoglou A+ C+ G+ T+ A- C- G- T- 1–p A model of CpG Islands – (2) Transitions Right now, aA+A+ + aA+C+ + aA+G+ + aA+T+ = 1 We need to adjust aij so as to allow transitions between (+) and (-) states Say we want with probability p++ to stay within CpG, p-- to stay within non-CpG 1. Let’s adjust all probs by that factor: example, let aA+G+ ← p++ × aA+G+ 2. Now, let’s calculate probs between (+) and (-) states 1. Total prob aA+S where S is a (-) state, is (1 – p++) 2. Let qA-, qC-, qG-, qT- be the proportions of A, C, G, and T within non-CpG states in training set 3. Then, let aA+A- = (1 – p++) × qA-; aA+C- = (1 – p++) × qC-; … 4. Do the same for (-) to (+) states 3. OK, but how do we estimate p++ and p--? 1. Estimate average length of a CpG island: l+ = 1/(1-p) ⇒ p = 1 – 1/l+ 2. Do the same for length between two CpG islands, l
- 12. CS262 Lecture 6, Win06, Batzoglou Applications of the model Given a DNA region x, The Viterbi algorithm predicts locations of CpG islands Given a nucleotide xi, (say xi = A) The Viterbi parse tells whether xi is in a CpG island in the most likely general scenario The Forward/Backward algorithms can calculate P(xi is in CpG island) = P(πi = A+ | x) Posterior Decoding can assign locally optimal predictions of CpG islands π^ i = argmaxk P(πi = k | x)
- 13. CS262 Lecture 6, Win06, Batzoglou What if a new genome comes? • We just sequenced the porcupine genome • We know CpG islands play the same role in this genome • However, we have no known CpG islands for porcupines • We suspect the frequency and characteristics of CpG islands are quite different in porcupines How do we adjust the parameters in our model? LEARNING
- 14. CS262 Lecture 6, Win06, Batzoglou Problem 3: Learning Re-estimate the parameters of the model based on training data
- 15. CS262 Lecture 6, Win06, Batzoglou Two learning scenarios 1. Estimation when the “right answer” is known Examples: GIVEN: a genomic region x = x1…x1,000,000 where we have good (experimental) annotations of the CpG islands GIVEN: the casino player allows us to observe him one evening, as he changes dice and produces 10,000 rolls 2. Estimation when the “right answer” is unknown Examples: GIVEN: the porcupine genome; we don’t know how frequent are the CpG islands there, neither do we know their composition GIVEN: 10,000 rolls of the casino player, but we don’t see when he changes dice QUESTION: Update the parameters θ of the model to maximize P(x|θ)
- 16. CS262 Lecture 6, Win06, Batzoglou 1. When the right answer is known Given x = x1…xN for which the true π = π1…πN is known, Define: Akl = # times k→l transition occurs in π Ek(b) = # times state k in π emits b in x We can show that the maximum likelihood parameters θ (maximize P(x|θ)) are: Akl Ek(b) akl = ––––– ek(b) = ––––––– Σi Aki Σc Ek(c)
- 17. CS262 Lecture 6, Win06, Batzoglou 1. When the right answer is known Intuition: When we know the underlying states, Best estimate is the average frequency of transitions & emissions that occur in the training data Drawback: Given little data, there may be overfitting: P(x|θ) is maximized, but θ is unreasonable 0 probabilities – VERY BAD Example: Given 10 casino rolls, we observe x = 2, 1, 5, 6, 1, 2, 3, 6, 2, 3 π = F, F, F, F, F, F, F, F, F, F Then: aFF = 1; aFL = 0 eF(1) = eF(3) = .2; eF(2) = .3; eF(4) = 0; eF(5) = eF(6) = .1
- 18. CS262 Lecture 6, Win06, Batzoglou Pseudocounts Solution for small training sets: Add pseudocounts Akl = # times k→l transition occurs in π + rkl Ek(b) = # times state k in π emits b in x + rk(b) rkl, rk(b) are pseudocounts representing our prior belief Larger pseudocounts ⇒ Strong priof belief Small pseudocounts (ε < 1): just to avoid 0 probabilities
- 19. CS262 Lecture 6, Win06, Batzoglou Pseudocounts Example: dishonest casino We will observe player for one day, 600 rolls Reasonable pseudocounts: r0F = r0L = rF0 = rL0 = 1; rFL = rLF = rFF = rLL = 1; rF(1) = rF(2) = … = rF(6) = 20 (strong belief fair is fair) rL(1) = rL(2) = … = rL(6) = 5 (wait and see for loaded) Above #s pretty arbitrary – assigning priors is an art
- 20. CS262 Lecture 6, Win06, Batzoglou 2. When the right answer is unknown We don’t know the true Akl, Ek(b) Idea: • We estimate our “best guess” on what Akl, Ek(b) are • We update the parameters of the model, based on our guess • We repeat
- 21. CS262 Lecture 6, Win06, Batzoglou 2. When the right answer is unknown Starting with our best guess of a model M, parameters θ: Given x = x1…xN for which the true π = π1…πN is unknown, We can get to a provably more likely parameter set θ i.e., θ that increases the probability P(x | θ) Principle: EXPECTATION MAXIMIZATION 1. Estimate Akl, Ek(b) in the training data 2. Update θ according to Akl, Ek(b) 3. Repeat 1 & 2, until convergence
- 22. CS262 Lecture 6, Win06, Batzoglou Estimating new parameters To estimate Akl: (assume | θCURRENT, in all formulas below) At each position i of sequence x, find probability transition k→l is used: P(πi = k, πi+1 = l | x) = [1/P(x)] × P(πi = k, πi+1 = l, x1…xN) = Q/P(x) where Q = P(x1…xi, πi = k, πi+1 = l, xi+1…xN) = = P(πi+1 = l, xi+1…xN | πi = k) P(x1…xi, πi = k) = = P(πi+1 = l, xi+1xi+2…xN | πi = k) fk(i) = = P(xi+2…xN | πi+1 = l) P(xi+1 | πi+1 = l) P(πi+1 = l | πi = k) fk(i) = = bl(i+1) el(xi+1) akl fk(i) fk(i) akl el(xi+1) bl(i+1) So: P(πi = k, πi+1 = l | x, θ) = –––––––––––––––––– P(x | θCURRENT)
- 23. CS262 Lecture 6, Win06, Batzoglou Estimating new parameters • So, Akl is the E[# times transition k→l, given current θ] fk(i) akl el(xi+1) bl(i+1) Akl = Σi P(πi = k, πi+1 = l | x, θ) = Σi ––––––––––––––––– P(x | θ) • Similarly, Ek(b) = [1/P(x | θ)]Σ{i | xi = b} fk(i) bk(i) k l xi+1 akl el(xi+1) bl(i+1)fk(i) x1………xi-1 xi+2………xN xi
- 24. CS262 Lecture 6, Win06, Batzoglou The Baum-Welch Algorithm Initialization: Pick the best-guess for model parameters (or arbitrary) Iteration: 1. Forward 2. Backward 3. Calculate Akl, Ek(b), given θCURRENT 4. Calculate new model parameters θNEW : akl, ek(b) 5. Calculate new log-likelihood P(x | θNEW) GUARANTEED TO BE HIGHER BY EXPECTATION-MAXIMIZATION Until P(x | θ) does not change much
- 25. CS262 Lecture 6, Win06, Batzoglou The Baum-Welch Algorithm Time Complexity: # iterations × O(K2 N) • Guaranteed to increase the log likelihood P(x | θ) • Not guaranteed to find globally best parameters Converges to local optimum, depending on initial conditions • Too many parameters / too large model: Overtraining
- 26. CS262 Lecture 6, Win06, Batzoglou Alternative: Viterbi Training Initialization: Same Iteration: 1. Perform Viterbi, to find π* 2. Calculate Akl, Ek(b) according to π* + pseudocounts 3. Calculate the new parameters akl, ek(b) Until convergence Notes: Not guaranteed to increase P(x | θ) Guaranteed to increase P(x, | θ, π* ) In general, worse performance than Baum-Welch
- 27. CS262 Lecture 6, Win06, Batzoglou Variants of HMMs
- 28. CS262 Lecture 6, Win06, Batzoglou Higher-order HMMs • How do we model “memory” larger than one time point? • P(πi+1 = l | πi = k) akl • P(πi+1 = l | πi = k, πi -1 = j) ajkl • … • A second order HMM with K states is equivalent to a first order HMM with K2 states state H state T aHT(prev = H) aHT(prev = T) aTH(prev = H) aTH(prev = T) state HH state HT state TH state TT aHHT aTTH aHTTaTHH aTHT aHTH
- 29. CS262 Lecture 6, Win06, Batzoglou Modeling the Duration of States Length distribution of region X: E[lX] = 1/(1-p) • Geometric distribution, with mean 1/(1-p) This is a significant disadvantage of HMMs Several solutions exist for modeling different length distributions X Y 1-p 1-q p q
- 30. CS262 Lecture 6, Win06, Batzoglou Example: exon lengths in genes
- 31. CS262 Lecture 6, Win06, Batzoglou Solution 1: Chain several states X Y 1-p 1-q p qXX Disadvantage: Still very inflexible lX = C + geometric with mean 1/(1-p)
- 32. CS262 Lecture 6, Win06, Batzoglou Solution 2: Negative binomial distribution Duration in X: m turns, where During first m – 1 turns, exactly n – 1 arrows to next state are followed During mth turn, an arrow to next state is followed m – 1 m – 1 P(lX = m) = n – 1 (1 – p)n-1+1 p(m-1)-(n-1) = n – 1 (1 – p)n pm-n X(n) p X(2)X(1) p 1 – p1 – p p …… Y 1 – p
- 33. CS262 Lecture 6, Win06, Batzoglou Example: genes in prokaryotes • EasyGene: Prokaryotic gene-finder Larsen TS, Krogh A • Negative binomial with n = 3
- 34. CS262 Lecture 6, Win06, Batzoglou Solution 3: Duration modeling Upon entering a state: 1. Choose duration d, according to probability distribution 2. Generate d letters according to emission probs 3. Take a transition to next state according to transition probs Disadvantage: Increase in complexity: Time: O(D) Space: O(1) where D = maximum duration of state F d<Df xi…xi+d-1 Pf Warning, Rabiner’s tutorial claims O(D2) & O(D) increases
- 35. CS262 Lecture 6, Win06, Batzoglou Viterbi with duration modeling Recall original iteration: VF(i) = maxk Vk(i – 1) akl × eF(xi) New iteration: VF(i) = maxk maxd=1…Df Vk(i – d) × Pf(d) × akF × Πj=i-d+1…ieF(xj) F L transitions emissions d<Df xi…xi + d – 1 emissions d<Dl xj…xj + d – 1 Pf Pl Precompute cumulative values
Hinweis der Redaktion
- From Wikipedia: DNA methylation is a type of chemical modification of DNA that can be inherited without changing the DNA sequence. As such, it is part of the epigenetic code. DNA methylation involves the addition of a methyl group to DNA — for example, to the number 5 carbon of the cytosine pyrimidine ring. DNA methylation is probably universal in eukaryotes . In humans , approximately 1% of DNA bases undergo DNA methylation. In adult somatic tissues, DNA methylation typically occurs in a CpG dinucleotide context; non-CpG methylation is prevalent in embryonic stem cells . In plants, cytosines are methylated both symmetrically (CpG or CpNpG) and asymmetrically (CpNpNp), where N can be any nucleotide. DNA methylation in mammals Between 60-70% of all CpGs are methylated. Unmethylated CpGs are grouped in clusters called " CpG islands " that are present in the 5' regulatory regions of many genes . In many disease processes such as cancer , gene promoter CpG islands acquire abnormal hypermethylation, which results in heritable transcriptional silencing. Reinforcement of the transcriptionally silent state is mediated by proteins that can bind methylated CpGs. These proteins, which are called methyl-CpG binding proteins , recruit histone deacetylases and other chromatin remodelling proteins that can modify histones, thereby forming compact, inactive chromatin termed heterochromatin . This link between DNA methylation and chromatin structure is very important. In particular, loss of Methyl-CpG-binding Protein 2 (MeCP2) has been implicated in Rett syndrome and Methyl-CpG binding domain protein 2 (MBD2) mediates the transcriptional silencing of hypermethylated genes in cancer. [ edit ] DNA methylation in humans In humans, the process of DNA methylation is carried out by three enzymes, DNA methyltransferase 1, 3a, and 3b (DNMT1, DNMT3a, DNMT3b). It is thought that DNMT3a and DNMT3b are the de novo methyltransferases that set up DNA methylation patterns early in development. DNMT1 is the proposed maintenance methyltransferase that is responsible for copying DNA methylation patterns to the daughter strands during DNA replication. DNMT3L is a protein that is homologous to the other DNMTs but has no catalytic activity. Instead, DNMT3L assists the de novo methyltransferases by increasing their ability to bind to DNA and stimulating their activity. Since many tumor suppressor genes are silenced by DNA methylation during carcinogenesis, there have been attempts to re-express these genes by inhibiting the DNMTs. 5-aza-2'-deoxycytidine (decitabine) is a nucleoside analog that inhibits DNMTs by trapping them in a covalent complex on DNA by preventing the ß-elimination step of catalysis, thus resulting in the enzymes' degradation. However, for decitabine to be active, it must be incorporated into the genome of the cell, but this can cause mutations in the daughter cells if the cell does not die. Additionally, decitabine is toxic to the bone marrow, a fact which limits the size of its therapeutic window. These pitfalls have led to the development of antisense RNA therapies that target the DNMTs by degrading their mRNAs and preventing their translation. However, it is currently unclear if targeting DNMT1 alone is sufficient to reactivate tumor suppressor genes silenced by DNA methylation. [edit] DNA methylation in plants Significant progress has been made in understanding DNA methylation in plants, specifically in the model plant, Arabidopsis thaliana . The principal DNA methyltransferases in A. thaliana , Met1, Cmt3, and Drm2, are similar at a sequence level to the mammalian methyltransferases. Drm2 is thought to participate in de-novo DNA methylation as well as in the maintenance of DNA methylation. Cmt3 and Met1 act principally in the maintenance of DNA methylation [1]. Other DNA methyltransferases are expressed in plants but have no known function (see [2]). The specificity for DNA methyltransferases is thought to be driven by RNA-directed DNA methylation. Specific RNA transcripts are produced from a genomic DNA template. These RNA transcripts may form double-stranded RNA molecules. The double stranded RNAs, through either the small interfering RNA (siRNA) or micro RNA (miRNA) pathways, direct the localization of DNA methyltransferases to specific targets in the genome [3]
- Why the algorithm has to be generalized is because in a standard HMM the output in each step would be one base, leading to state durations of geometric length. If we look at empirical data, such as these plots, we see that the intron lengths seem to follow the geometric distribution fairly well, but for the exon that would be a pretty bad model. So in our state space the exons are generalized states, choosing a length from a general distribution and outputting the entire exon in one step, while the intron and intergene states still output one base at a time and follow the geometric distribution.