2. Component Processes
Absorption – entry of a drug from its site
of administration to the systemic
circulation
Distribution – process by which a drug
enters the interstitium or tissues from the
blood
Metabolism / Biotransformation –
processes by which a drug is changed: to
its active form or to its removable form
Excretion – removal of the drug from the
body
3. Drug
ABSORPTION into Plasma
DISTRIBUTION
to Tissues
Bound Drug
Free Drug
Tissue
Storage
Sites of
Action
Drug METABOLISM:
Liver, Lung, etc
Drug EXCRETION:
Renal, Biliary, etc.
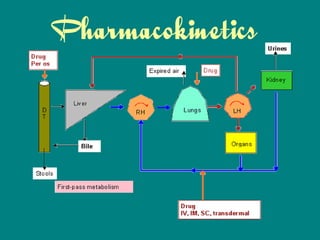
Drug Biodisposition / Pharmacokinetics
4. Permeation
Permeation – travel of a drug across
cellular membranes, influencing its
biodisposition; is dependent on:
Solubility
Ionization
Concentration gradient
Surface area
Tissue vascularity
5. Drug Permeation
Solubility
Lipid solubility – ability to pass through lipid bilayers
Water solubility – in aqueous phases
Partition coefficient – ratio of lipid to aqueous solubility : the higher
the partition coeff, the more membrane soluble the drug
Ionization
The Henderson–Hasselbach equation – determines the percentage
of ionization (ionized=water-soluble; nonionized=lipid-soluble)
Drugs are either weak acids or weak bases, & can exist as charged
or neutral particles in equilibrium, depending on pH & pKa
Ionization increases renal clearance of drugs
6. Drug Permeation
Concentration gradient – diffusion is down a
concentration gradient; the greater the
concentration gradient, the faster the
diffusion/permeation
Surface area – the available area for
permeation; the greater the surface area, the
faster the diffusion / permeation
Tissue Vascularity – density of blood supply &
speed of blood flow – the better/more the tissue
vascularity, the better the permeation
7. Absorption
Passive diffusion – most common
Aqueous diffusion: Fick’s Law:
Flux (J) = (C1 – C2) x S.A. x P.
coefficient
Thickness
J = molecules per unit time
C1= higher concentration
C2 = lower concentration
S.A. = surface area available for diffusion
P. Coefficient = permeability coefficient / partition coefficient
Thickness = length of the diffusion path
9. weak Acids & weak Bases
A weak acid is a neutral molecule that dissociates into
an anion & a proton (H+) so that its protonated form is
neutral, more lipid-soluble
A weak base is a neutral molecule that can form a cation
by combining with a proton so its protonated form is
charged, water-soluble
weak acids pKa weak bases pKa
Phenobarbital 7.1 Cocaine 8.5
Pentobarbital 8.1 Ephedrine 9.6
Acetaminophen 9.5 Chlordiazepoxide 4.6
Aspirin 3.5 Morphine 7.9
10. Diffusion
Aqueous diffusion
within large aqueous
compartments
across tight junctions
across endothelium thru
pores (MW20,000 - 30,000)
molecules tend to move
from an area of higher to an
area of lower concentration
plasma protein-bound drugs
cannot permeate thru
aqueous pores
charged drugs will be
influenced by electric fields
Lipid diffusion
higher partition coefficient =
easier for a drug to enter lipid
phase from aqueous
charged drugs – difficulty in
diffusing thru lipid
uncharged – lipid-soluble
lower pH relative to pKa,
greater fraction of protonated
drug (protonated form of an
acid is neutral; protonated
form of a base is charged)
A weak acid at acid pH & a
weak base at alkaline pH will
be more lipid-soluble
11. Carrier – mediated Transport
Facilitated diffusion – passive (no E
expended) carrier-mediated transport.
saturable;
subject to competitive & non-competitive inhibition
used by peptides, amino acids, glucose
Active (uses E) carrier-mediated transport
saturable
subject to competitive & non-competitive inhibition
against a concentration gradient
e.g. Na – K pump
12. Endocytosis & Exocytosis
ENDOCYTOSIS
entry into cells by very large substances (uses
E)
e.g. Iron & vit B12 complexed with their
binding proteins into intestinal mucosal cells
EXOCYTOSIS
expulsion of substances from the cells into the
ECF (uses E)
e.g. Neurotransmitters at the synaptic junction
13. Ion Trapping
Ion trapping or reabsorption – delays excretion
Kidneys:
nearly all drugs are filtered at the glomerulus
most drugs in a lipid-soluble form will be reabsorbed by
passive diffusion
to increase excretion: change urinary pH to favor the charged
form of the drug (not readily absorbed)
– weak acids are excreted faster in alkaline pH (anion form favored)
– weak bases are excreted faster in acidic pH (cation form favored)
Other sites: body fluids where pH differs from blood pH, favoring
trapping or reabsorption
stomach contents ▪ aqueous humor
small intestines ▪ vaginal secretions
breast milk ▪ prostatic secretions
14.
15. Distribution
First pass effect – decreased bioavailability of
drugs administered orally because of initial
absorption into the portal circulation &
distribution in the liver where they may undergo
metabolism or excretion into bile
Extraction Ratio – magnitude of the first pass
effect.
ER = cl Liver / q (hepatic blood flow)
Systemic drug bioavailability – determined from
extent of absorption & ER.
F = f x (1 – ER)
16. Distribution
Volume of Distribution – ratio between the
amount of drug in the body (dose given) &
the concentration of the drug in blood
plasma. Vd = drug in body / drug in blood
Factors influencing Vd:
drug pKa (permeation)
extent of drug-plasma protein binding
lipid solubility (partition coefficient)
patient age, gender, disease states, body composition
17. Drug – Plasma Protein Binding
Most drugs are bound to some extent to plasma
proteins Albumin, Lipoproteins, alpha 1 acid
glycoprotein
Extent of protein binding parallels drug lipid
solubility
Binding of drug to Albumin is often non-
selective,
Acidophilic drugs bind to Albumin, basophilic
drugs bind to Globulins
drugs with similar chemical/physical properties
may compete for the same binding sites
Volume of distribution is inversely proportional to
protein binding
18. Distribution
Non-ionized (hydrophobic) drugs cross
biomembranes easily
Binding to plasma proteins accelerates
absorption into plasma but slows diffusion into
tissues
Unbound / free drug crosses biomembranes
Competition between drugs may lead to
displacement of a previously bound drug
higher levels of free/unbound drug better
distribution
Distribution occurs more rapidly with high blood
flow & high vessel permeability
19. Distribution
Special barriers to distribution:
placenta
blood-brain barrier
Many disease states alter distribution:
Edematous states – cirrhosis, heart failure, nephrotic syndrome –
prolong distribution & delay Clearance
Obesity allows for greater accumulation of lipophilic agents within
fat cells, increasing distribution & prolonging half-life
Pregnancy increases intravascular volume, thus increasing
distribution
hypoAlbuminemia allows drugs that normally bind to it to have
increased bioavailability
Renal failure may decrease drug bound fraction (metabolite
competes for protein binding sites) & thus ↑ free drug levels
20. Blood Brain Barrier (BBB):
Only lipid-soluble compounds get through the BBB.
Four components to the blood-brain barrier:
Tight Junctions in brain capillaries
Glial cell foot processes wrap around the capillaries
Low CSF protein concentration ------> no oncotic pressure for
reabsorbing protein out of the plasma.
Endothelial cells in the brain contain enzymes that metabolize,
neutralize, many drugs before they access the CSF.
– MAO and COMT are found in brain endothelial cells. They
metabolize Dopamine before it reaches the CSF, thus we
must give L-DOPA in order to get dopamine to the CSF.
21. Exceptions to the BBB. Certain parts of the brain are not
protected by the BBB:
Pituitary, Median Eminence
Supraventricular areas
Parts of hypothalamus
Meningitis: It opens up the blood brain barrier due to edema.
Thus Penicillin-G can be used to treat meningitis (caused by
Neisseria meningitides), despite the fact that it doesn't normally
cross the BBB. Penicillin-G is also actively pumped back out of
the brain once it has crossed the BBB.
Sites of Concentration: can affect the Vd
Fat, Bone, any Tissue, Transcellular sites: drug concentrates in
Fat / Bone / non-Plasma locations lower concentration of drug
in Plasma higher Vd
22.
23. Metabolism
Biotransformation of drugs (usually in the Liver; also in the
Lungs, Skin, Kidney, GIT)) to more polar, hydrophilic,
biologically inactive molecules; required for elimination
from the body.
Phase I reactions – alteration of the parent drug by
exposing a functional group; active drug transformed by
phase I reactions usually lose pharmacologic activity,
while inactive prodrugs are converted to biologically
active metabolites
Phase II reactions – parent drug undergoes conjugation
reactions (to make them more soluble) that form
covalent linkages with a functional group: glucuronic
acid, acetyl coA, sulfate, glutathione, amino acids,
acetate, S-adenosyl-methionine
24. Metabolism
Phase I
reaction products may be directly excreted in urine or
react with endogenous compounds to form water-soluble
conjugates
mixed function oxidase system (cytochrome P450
enzyme complex: Cyt P450 enzyme, Cyt P450
reductase) requires NADPH (not ATP) as E source, &
molecular O2; [drug metabolizing enzymes are located in
hepatic microsomes: lipophilic, endoplasmic reticulum
membranes (SER)]
Phase I enzymes perform multiple types of reactions:
OXIDATIVE REACTIONS
REDUCTIVE REACTIONS
HYDROLYTIC REACTIONS
25. CYTOCHROME-P450 COMPLEX:
There are multiple isotypes.
CYT-P450-2, CYT-P450-3A are responsible for the metabolism of most drugs.
CYT-P450-3A4 metabolizes many drugs in the GIT, decreasing the
bioavailability of many orally absorbed drugs.
INDUCERS of CYT-P450 COMPLEX: Drugs that
increase the production or ↓ degradation of Cyt-P450
enzymes.
Phenobarbital, Phenytoin, Carbamazepine induce CYT-P450-3A4
Phenobarbital, Phenytoin also induce CYT-P450-2B1
Polycyclic Aromatics (PAH): Induce CYT-P450-1A1
Glucocorticoids induce CYT-P450-3A4
Chronic Alcoholism, Isoniazid induce CYT-P450-2E1. important! this drug
activates some carcinogens e.g. Nitrosamines.
*Chronic alcoholics have up-regulated many of their CYT-P450 enzymes.
26. INHIBITORS of CYT-P450 COMPLEX
Inhibit production: Ethanol suppresses many of the CYT-P450
enzymes, explaining some of the drug-interactions of acute
alcohol use.
Non–competitive inhibition: Chloramphenicol is metabolized by
Cyt P450 to an alkylating metabolite that inactivates Cyt P450
Competitive inhibition: Erythromycin inhibits CYT-P450-3A4.
Terfenadine (Seldane) is metabolized by CYT-P450-3A4, so the
toxic unmetabolized form builds up in the presence of
Erythromycin. The unmetabolized form is toxic and causes lethal
arrhythmias. This is why Seldane was taken off the market;
Cimetidine, Ketoconazole – bind to the heme in Cyt P450,
decreasing metabolism of Testosterone & other drugs
Steroids: Ethinyl estradiol, Norethindrone; Spironolactone;
Propylthiouracil (PTU): inactivate Cyt P450 by binding the heme
27. Metabolism
Phase II
Drug Conjugation reactions: “detoxification” rxns: non-
microsomal, primarily in the liver; also in plasma & GIT –
usually to glucuronides, making the drug more soluble.
conjugates are highly polar, generally biologically
inactive (exception: morphine glucuronide – more potent
analgesic than the parent compound) & tend to be
rapidly excreted in urine or bile
“Enterohepatic recirculation”: high molecular weight
conjugates are more likely to be excreted in bile
intestines, where N flora cleave the conjugate bonds,
releasing the parent compound into the systemic
circulation delayed parent drug elimination &
prolongation of drug effects
conjugation, hydrolysis, oxidation, reduction
30. Toxicity
drugs are metabolized to toxic products
hepatotoxicity exhibited by
acyl glucuronidation of NSAIDS
N-acetylation of Isoniazid
Acetaminophen in high doses – glucuronidation &
sulfation are usual conjugation reactions in therapeutic
doses, but in high doses, these get saturated so Cyt
P450 metabolizes the drug, forming hepatotoxic reactive
electrophilic metabolites fulminant hepatotoxicity &
death (antidote: N-acetylcysteine)
31. Reduction in Bioavailability
First pass effect
Intestinal flora metabolize the drug
Drug is unstable in gastric acid e.g.
Penicillin
Drug is metabolized by digestive enzymes
e.g. Insulin
Drug is metabolized by intestinal wall
enzymes e.g. sympathomimetic drugs /
catecholamines
32.
33. Excretion
Clearance – CL – removal of drug from the
blood, or the amount of blood/plasma that is
completely freed of drug per unit time over the
plasma concentration of the drug
CL = rate of elimination of drug
plasma drug concentration
especially important for ensuring appropriate long-term dosing, or
maintaining correct steady state drug concentrations
Renal clearance - unchanged drug, water-soluble metabolites –
glomerular filtration, active tubular secretion, passive tubular
reabsorption of lipid-soluble agents
Hepatic clearance – extraction of drugs after GIT absorption
34. Excretion
KIDNEY
GLOMERULAR FILTRATION: Clearance of the apparent volume
of distribution by passive filtration.
Drug with MW < 5000 ------> it is completely filtered.
Inulin is completely filtered, and its clearance can be
measured to estimate Glomerular Filtration Rate (GFR).
TUBULAR SECRETION: Active secretion.
Specific Compounds that are secreted:
– para-Amino Hippurate (PAH) is completely secreted, so its
clearance can be measured to estimate Renal Blood Flow
(RBF).
– Penicillin-G is excreted by active secretion. Probenecid can
be given to block this secretion.
35. Excretion
Half life (t ½) – time required to decrease the
amount of drug in the body by 50% during
elimination or during a constant infusion; useful
in
estimating time to steady-state: approximately 4 half-lives to
reach 94%
Estimation of time required for drug removal from the body
Estimation of appropriate dosing interval: drug accumulation
occurs when dosing interval is less than 4 half-lives
Affected by
Chronic renal failure – decreases clearance, prolongs half-life
increasing Age – Vd changes, prolongs half-life
Decreased plasma protein binding shortens half-life
36. Half – Life
The half-life is inversely proportional to the
Kel, constant of elimination. The higher
the elimination constant, the shorter the
half-life.
37. Drug Elimination
Zero order kinetics – rate of elimination of the
drug is constant regardless of concentration i.e.
constant amount of drug eliminated per unit time
so that concentration decreases linearly with
time
examples: ethanol, phenytoin, aspirin
First order kinetics – rate of elimination of the
drug proportional to concentration i.e. constant
fraction of the drug eliminated per unit time so
that concentration decreases exponentially over
time