

Empfohlen
Weitere ähnliche Inhalte
Was ist angesagt?
Was ist angesagt? (20)
Ähnlich wie Minarcik robbins 2013_ch14-wbc
Ähnlich wie Minarcik robbins 2013_ch14-wbc (20)
Mehr von Elsa von Licy
Mehr von Elsa von Licy (20)
Kürzlich hochgeladen
Kürzlich hochgeladen (20)
Minarcik robbins 2013_ch14-wbc
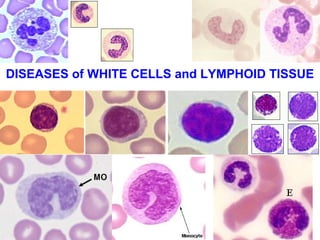
- 1. DISEASES of WHITE CELLS and LYMPHOID TISSUE
- 2. • • • • • • • • • • • • • • Topics for Chapter 14 Leukopenia/Neutropenia Leukocytosis Lymphadenitis/Lymphadenopathy (Malignant) Lymphoma NON-Hodgkins Lymphoma Hodgkins Lymphoma (Hodgkins Disease) ALL/CLL (Acute/Chronic Lymphocytic Leukemia) Multiple Myeloma M1/M2/M3/M4/M5/M6/M7 Myeloproliferative Disorder CML and Polycythemia Vera Essential Thrombocytosis Splenomegaly Thymoma
- 3. WBC/LYMPHOID DISORDERS • • • • • • Review of Normal WBC Structure/Function Benign Neutrophil and Lymphoid Disorders Leukemias Lymph Nodes Spleen/Thymus REVIEW
- 5. NEUTROPHILS • Normal TOTAL WBC count 6-11 K • Neutrophils usually 2/3 of total normal • Myeloblast Promyelocyte Myelocyte Metamyelocyte Band (stab) Mature Neutrophil (Poly, PMN, Neutrophilic Granulocyte) • Produced in red (hematopoetic) marrow, sequester (pool) in spleen, live in peripheral blood, migrate OUT of vascular compartment PRN, live a couple days normally
- 6. NEUTROPHIL Neutrophil Polymorphonuclear Leukocyte, PMN, PML “Leukocyte” Granulocyte, Neutrophilic granulocyte “Poly-” Polymorph
- 8. LYSOSOMAL CONSTITUENTS • PRIMARY • Also called AZUROPHILIC, or NON-specific • Myeloperoxidase • Lysozyme (anit-Bact.) • Acid Hydrolases • SECONDARY • • • • • Also called SPECIFIC Lactoferrin (anti-Bact.) Lysozyme (anti-Bact.) Alkaline Phosphatase Collagenase
- 9. • • • • • • FUNCTIONS Margination Rolling Adhesion Transmigration (Diapedesis) Chemotaxis Phagocytosis: RecognitionEngulfmentKilling (digestion) • Equilibrium with splenic pool
- 10. PELGER-HUET ANOMALY • Genetic: Autosomal Dominant) • Sometimes ACQUIRED (Pseudo-PELGER-HUET) • All neutrophils look like BANDS • NOT serious, mostly a cute incidental finding
- 11. CHEDIAK-HIGASHI SYNDROME • Also genetic: Autosomal Recessive • Abnormal LARGE irregular neutrophil granules • Impaired lysosomal digestion of bacteria • Associated with pigment and bleeding disorders • CAN be serious, especially in kids
- 12. LEUKO-penia/NEUTRO-penia Neutropenia/Agranulocytosis • INADEQUATE PRODUCTION • INCREASED DESTRUCTION • 500-1000/mm3 is the DANGER zone!
- 13. INADEQUATE PRODUCTION • Stem cell suppression, e.g., aplastic anemias • DRUGS, esp. CHEMO, MANY antibiotics, aminopyrene, thio-uracil, phenylbutazone • DNA suppression due to megaloblastic/myelodysplastic states • Kostmann Syndrome: (A-R) (genetic, congenital) • Marrow usually shows granulocytic HYPOplasia, just as in RBC and PLAT decreased production
- 14. INCREASED DESTRUCTION • Immune mediated – By itself (idiopathic), or as in SLE – After “sensitization” by many drugs • Splenic sequestration, hypersplenism • Increased peripheral demand, as in overwhelming infections, esp. fungal • Marrow usually shows granulocytic HYPER-plasia, just as in RBC and PLAT increased destructions
- 15. Leukocytosis/Neutrophilia • • • • Marrow and splenic pool size Rate of release between pool and circulation Marginating pool Rate of WBCs (neutrophils/monocytes) leaving the vascular compartment • NON-vascular pools FIFTY times larger than the vascular pools • TNF/IL-1/cytokines stimulate T-cells to produce CSF, the WBC equivalent of EPO
- 16. NEUTROPHIL INCREASES (e.g., “NEUTROPHILIA”) • BACTERIA • TISSUE NECROSIS, e.g., MI • DÖHLE BODIES and TOXIC GRANULES are often seen with NEUTROPHILIA • Accompanied by a “LEFT” shift
- 17. EOSINOPHIL INCREASES (i.e., “EOSINOPHILIA”) • ALLERGIES (esp. DRUG allergies) • PARASITES Is there such a thing as a specific eosinopenia? ANS: NO
- 18. BASOPHIL INCREASES (i.e., “BASOPHILIA”) • RARE. VERY RARE. Period. • But if you want to remember something at least, remember myeloproliferative diseases in which ALL cell lines are increased Is there such a thing as a specific baso-penia? ANS: NO
- 19. • TB • • • • • MONOCYTE INCREASES (i.e., “MONOCYTOSIS”) SBE RICKETTSIAL DISEASES MALARIA SLE IBD, i.e., ULCERATIVE COLITIS
- 20. LYMPHOCYTE INCREASES (i.e., “LYMPHOCYTOSIS”) • TB • VIRAL –Hep-A –CMV –EBV • Pertussis (whooping cough)
- 21. “MYELOPROLIFERATIVE” disorders • Also called “chronic” myeloproliferative disorders because they last for years • Differentiate: Myeloproliferative vs. Myelodysplastic • ALL marrow cell lines are affected, splenomegaly • Proliferating cells do NOT suppress residual marrow production, and go OUTSIDE marrow, and EXPAND marrow to fatty appendicular marrow • Associated with EXTRA-medullary hematopoesis – Chronic Myelogenous “Leukemia” (CML) – P. Vera
- 22. CML • NOT AT ALL like an “acute” leukemia, but can develop into an acute leukemia, as a condition called a “blast crisis” • Age: adult, NOT kids • 90% have the “Philadelphia” chromosome, which are aberrations on chromosome #9 (BCR) and #22 (ABL), the BCR-ABL “fusion”
- 23. CML • Marrow 100% cellular, NOT 50% • ALL cell lines increased, M:E ratio massively increased, 50K-100K neutrophils with SIGNIFICANT “left shift”, but not more than 10% blasts • SIGNIFICANT SPLENOMEGALY!!!!! • Significant breakthrough with BCR-ABL kinase inhibitors!!! (90% remissions)
- 26. Polycythemia Vera • All cell lines increased, NOT just RBC • HIGH marrow cell turnover stimulates increased purines which often cause gout (10%) • BOTH thrombosis AND bleeding risks are present because the increased platelets are AB-normal • Do not get “blast” crises, BUT can progress to myelofibrosis
- 28. ESSENTIAL THROMOCYTOSIS • Platelet count often near 1 million/mm3 • Often a diagnosis of exclusion. • The RAREST of all myeloproliferative disorders • Giant platelets usually. Why? Ans: Quicker release from marrow (RPW/RDW) (MPV/MCV) • Massively increased megakaryocytes in the marrow
- 30. PRIMARY MYELOFIBROSIS • • • • Rapid progressive marrow fibrosis Oldest age group of all the MPD’s, >60 Can follow other MPD’s. Why? Usually the most extensive extramedullary hematopoesis because the marrow is NOT the primary site of hematopoesis • LEUKOERYTHROBLASTOSIS • Like CML, 10-20% can progress to AML
- 32. WBC/LYMPHOID DISORDERS • Review of Normal WBC Structure/Function • Benign Neutrophil and Lymphoid Disorders • Leukemias • Lymph Nodes • Spleen/Thymus • REVIEW
- 33. LEUKEMIAS • MALIGNANT PROLIFERATIONS of WHITE BLOOD CALLS • In the case of neutrophilic precursors, the primary process is marrow and peripheral blood, but can involve any organ or tissue which receives blood • In the case of lymphocytes, there is an intimate concurrence with malignant lymphomas
- 34. Lymphocytic Leukemias vs. Lymphomas • All leukemias of lymphocytes have lymphoma counterparts • Primary lymphomas can have “leukemic” phases, including multiple myelomas • Any myeloid leukemia can infiltrate a lymph node, or any other site, but if/when it does it is NOT called a lymphoma, but simply a myeloid infiltrate INTO a lymph node • ALL lymphomas are malignant proliferations of lymphocytes • ALL leukemias involve bone marrow changes
- 35. • • • • • • LYMPHOMAS NODAL or EXTRANODAL T or B SMALL or LARGE CELLS FOLLICULAR or DIFFUSE Hodgkins or NON-Hodgkins “F.A.B. classification” is currently popular this week (FrenchAmericaBritish), for the NON-Hodgkins lymphomas, also evolved into the “International” classification
- 36. • • • • • LEUKEMIAS Acute or Chronic Myeloid or Lymphocytic Childhood or Adult All involve marrow All ACUTE leukemias suppress normal hematopoesis, i.e., have anemia, thrombocytopenia • Most have predictable chromosomal aberrations • Some can respond DRASTICALLY to chemo, most notably ALL in children, even be cured!!!!
- 37. BLAST
- 38. WHITE CELL NEOPLASMS Leuk/Lymph • Many have predictable chromosomal translocations • Can arise in inherited and/or genetic diseases: – Downs Syndrome (Trisomy 21) – Fanconi’s anemia (hereditary aplastic anemia) – Ataxia telangiectasia • May have a STRONG viral relationship: – HTLV-1 (lymphoid tumors) – EBV (Burkitt Lymphoma) – (in HIV): Human Herpesvirus-8 (KS) and B-Cell Lymphomas
- 39. WHITE CELL NEOPLASMS Leuk/Lymph • Can be caused by H. Pylori (gastric B-Cell lymphomas) • Can follow celiac disease (gluten sensitive enteropathy T-Cell lymphomas) • Are common in HIV, B-Cell lymphomas, CNS lymphomas
- 40. A.L.L./LYMPHOMAS* • SUDDEN ONSET • ANEMIA, BLEEDING, FEVER • Bone pain, adenopathy, hepatosplenomegaly • CNS: headaches, vomiting, nerve palsies * • ( NB: These are pretty much the clinical symptoms of A.M.L. too and vice versa)
- 41. A.L.L./LYMPHOMAS • “Lymphoblasts” which can give rise either to T or B cells are the cells of malignant proliferation • All lymphocytic leukemias CANNOT be classified independently of lymphomas because they all have lymphoma counterparts • A.L.L. mostly in children • Most have chromosomal changes, hyperploidy, Philadelphia chromosome, translocations • SIGNIFICANT response to chemo: 90% remission, 75% CURE!!!
- 42. A.L.L.
- 43. C.L.L. • Unexplained sustained (months) lymph count of > 4000/mm3 is CLL, usually picked up on CBC • M>F, age >60 • Lymphs look normal and are NOT blasts • No need for marrow exam for dx, but progressive involvement of marrow, nodes, and other organs is the usual biologic behavior • Liver can be involved portally or sinusoidally • Translocations RARE, but trisomies and deletions common
- 44. C.L.L.
- 45. C.L.L. • HYPO-gammaglobulinemia • 15% have antibodies against RBC’s or PLATS • CANNOT be classified as separate from lymphomas
- 46. MULTIPLE MYELOMA • DEFINED AS A MALIGNANT PROLIFERATION OF PLASMA CELLS (i.e., former B-lymphocytes) • Can have a “leukemic” phase, but the BONE MARROW is the usual primary site of origin • Usually have MONOCLONAL GAMMOPATHIES • Secrete Heavy and Light chains, and Light chains in the urine is known as Bence-Jones protein • Usually have elevated IL-6 (bad prognosis)
- 47. PLASMA CELL classic features • OVAL cytoplasm, ROUND nucleus off to side • Cartwheel/Clockface chromatin • Prominent Golgi or “Hoff”
- 48. MONOCLONAL “SPIKE” on SPE NORMAL MULTIPLE MYELOMA
- 49. MULTIPLE MYELOMA • BONE DESTRUCTION • Various deletions and translocations • Plasma cells usually 1-3% of marrow, but >20% or plasma cells in SHEETS is diagnostic • Plasma cells usually look normal • IgG >> IgA, other immunoglobulins are rare • Staph, Strep, E. coli infections • Bleeding* • Amyloidosis • RENAL FAILURE
- 50. Multiple Myeloma: Skull X-ray
- 51. “Solitary” Plasmacytoma • Progression to MM is “inevitable”, with time, perhaps 10-20 years even
- 52. M.G.U.S. • Monoclonal Gammopathy of Unknown Significance, i.e., no plasma cell proliferation is found • Age related • 1% of 50-year olds, 3% of 70-year olds, etc. • Same chromosomal aberrations as MM, but generally follow a BENIGN course
- 53. Other “GAMMOPATHIES” • Waldenstrom’s MACROglobulinemia IgM (associated with lymphomas) • Heavy Chain Disease (associated with lymphomas) • AMYLOID, follows MM and/or chronic granulomatous diseases
- 54. • • • • A.M.L. GENETIC ABERRATIONS INHIBIT DIFFERENTIATION Many have various TRANSLOCATIONS F.A.B. classifies them as M0 M7 MORE than 20% of BLASTS are needed in the marrow for a diagnosis of acute leukemia!!! (i.e., ANY kind of BLAST • NORMALLY, a marrow should have only about 1-2 % blasts
- 55. • M0 A.M.L. Minimally differentiated • M1 AUER rods rare • M2 AUER rods common • M3 (COMMON) (COMMON) Acute PRO-myelocytic leukemia • M4 AMML (myelo-Mono cytic) (COMMON) • M5 • M6 • M7 Monocytic ErythroLeukemia Acute Megakaryocytic leukemia NOTE: Diagnosis is CONFIRMED by special markers, not just visual identification
- 56. M0M2
- 57. M3
- 60. A.M.L. • Anemia • Thrombocytopenia (bleeding) – Petechiae – Ecchymoses • • • • Fever Fatigue Lymphadenopathy 60% respond, BUT only 20 % are free of remission after 5 years, WORSE than A.L.L.
- 61. MYELO-DYSPLASTIC SYNDROMES • Increased risk of acute leukemias • But, UNLIKE the myeloPROLIFERATIVE syndromes, NOT a hypercellular marrow • Spontaneous or drug related (even > 5 yrs!) • Has marrow ABERRATIONS – REFRACTORY ANEMIAS – RINGED SIDEROBLASTS (Fe in mitochondria) – Nuclear “BUDDING” – EXCESS BLASTS, but LESS than 20% – About, say, 25% develop into acute leukemias
- 62. Ring Sideroblasts and “BUDS”
- 63. LYMPH NODES • Normal Structure, Function • Benign enlargement/Benign disease – Acute – Chronic (follicular vs. “sinus histiocytosis”) • Lymphomas/Malignant Lymphomas – Adjectives of various classifications – Features – STAGING • Metastatic disease TO lymph nodes
- 65. Blood flow? Lymph flow? CORTEX ---SUB-capsular Sinus ---Follicles (Pri? Or second.?) ---PARA-follicular zone MEDULLA
- 66. • • • • • • • • • • Definition of TERMS Lymphadenopathy Lymphadenitis Dermatopathic Normal size? Palpation What to do if a lymph node is enlarged? Diffuse/Follicular T/B/NK, Small/Large, Cleaved/Non-cleaved Precursor/Peripheral HD/Non-HD
- 67. BENIGN ENLARGEMENT • • • • Also called LYMPHADENITIS, and HYPERPLASIA Can be ACUTE (tender), or CHRONIC (non-tender) Usually SUBSIDE in, say, less than 6 weeks FOLLICULAR HYPERPLASIA is enlargement of the cortical secondary follicles and increase in number of the cortical secondary follicles • SINUS HISTIOCYTOSIS is prominence in medullary sinuses (also called “reticular” hyperplasia)
- 70. (MALIGNANT) LYMPHOMAS • Terms in historic classifications: – – – – Diffuse/Follicular, Small/Large, Cleaved/Non-cleaved Hodgkins (REED-STERNBERG CELL) /NON-Hodgkins Lukes, Rappaport, etc. Working Formulation, WHO, NIH, FAB, Intl., etc. –B –T – PRECURSOR (less mature looking) – PERIPHERAL (more mature looking)
- 71. DIFFUSE LYMPHOMA
- 77. FEATURES of LYMPHOMAS • The antigen receptor genes re-arrangement PRECEDES malignant transformation, so the cells are MONOCLONAL, NOT the usual POLYCLONAL • 85% B-cell, 15% T-Cell • The tumor cells congregate wherever T and B cell congregate normally however • DISRUPTED or “EFFACED” normal architecture, obliterated subcapsular sinus • HD/Non-HD staging CRUCIALLY IMPORTANT, esp. HD. Why? HD grows (spreads) more “linearly”, i.e., more “predictably”.
- 78. LATEST CLASSIFICATION • NON-HODGKIN – PRECURSOR B – PERIPHERAL B – PRECURSOR T – PERIPHERAL T • HODGKIN’S DISEASE (i.e., HODGKINS LYMPHOMA) NS, LP, MC, LD
- 79. PRECURSOR B • Precursor B LYMPHOBLASTIC LEUKEMIA/LYMPHOMA
- 80. PERIPHERAL B • CHRONIC LYMPHOCYTIC LEUKEMIA/LYMPHOMA • • • • • • • • • • • B-Cell PRO-lymphocytic LEUKEMIA Lymphoplasmacytic Splenic and Nodal Marginal Zone EXTRA-nodal Marginal Zone Mantle Cell Follicular Marginal Zone Hairy Cell Leukemia Plasmacytoma/Multiple Myeloma Diffuse B Cell BURKITT LYMPHOMA (Starry Sky)
- 81. PRECURSOR T • Precursor T LYMPHOBLASTIC LEUKEMIA/LYMPHOMA
- 82. • • • • • • • • • • • • PERIPHERAL T and NK T-Cell PRO-Lymphocytic Leukemia Large Granular Mycossis fungoides/Sezary Cell syndrome (skin) Peripheral T-Cell Anaplastic large cell Angioimmunoblastic T-Cell Enteropathy-associated T-Cell Panniculitis-like Hepatosplenic gamma-delta Adult T-Cell NK/T Cell nasal NK-Cell leukemia
- 83. • • • • • • • LYMPHOCYTE MARKERS (CD-) i.e., LYMPHOCYTE ANTIGENS T-Cell: 1,3,4,5,8 B-Cell: 10 (CALLA), 19,20,21,23,79a Mono/Mac: 11c, 13, 14, 15, 33, 34 STEM: 34 RS: 15, 30 All: 45 (Leukocyte Common Antigen) NK: (16, 56)
- 84. HODGKINS DISEASE • NEED R-S (Reed-Sternberg, or Sternberg-Reed) cells for correct diagnosis –NODULAR SCLEROSIS (Young Women), the R-S cells may be called “LACUNAR” cells –MIXED CELLULARITY – Lymphocyte RICH – Lymphocyte POOR – Lymphocyte PREDOMINANCE
- 86. STAGING, HD & NHD • I ONE NODE or NODE GROUP • II MORE than ONE, but on ONE side of diaph. • III BOTH sides of diaph., but still in nodes only • IV OUTSIDE of NODES, e.g., liver, marrow, etc. • A • B No systemic symptoms fever and/or night sweats and/or 10% weight loss
- 87. METASTATIC CARCINOMA • Perhaps the single most important staging and prognostic feature of tumors • The metastatic cells FIRST enter into the SUBCAPSULAR SINUS • The tumor may replace the entire node and enlarge it • The tumor may be focal • The tumor usually looks the same as it’s primary or other metastases • The tumor usually ENLARGES the node
- 91. SPLEEN • 150 grams POST-LUQ (just like kidney, 1/10 of liver) • Bordered by diaphragm, kidney, pancreas, splenic flexure, stomach • SMOOTH & GLISTENING capsule • ~~~50% RED pulp, 50% WHITE pulp
- 93. ABNORMAL SPLEEN
- 94. ABNORMAL SPLEEN
- 95. SPLENIC FUNCTION • REMOVE OLD BLOOD CELLS • MAJOR SECONDARY ORGAN of the IMMUNE SYSTEM • HEMATOPOIESIS • SEQUESTER (POOL) BLOOD CELLS • 15% of body’s PHAGOCYTIC activity is in the spleen (liver has >80)
- 96. SPLENOMEGALY • CONGESTIVE vs INFILTRATIVE • HYPERSPLENISM –Anemia –Leukopenia –Thrombocytopenia • DECISION for SPLENECTOMY
- 97. SPLENOMEGALY • INFECTIONS: TB, Mono, Malaria, Fungus • PORTAL HTN: CHF, CIRRHOSIS, PV Thromb. • LYMPHOHEMATOGENOUS: Leuk, Lymph, esp. CML • IMMUNE: RA, SLE • STORAGE: Gaucher, Niemann-Pick • MISC: Amyloid, mets (melanoma, lymphoma, germ cell tumors of testis) LONG STANDING CONGESTION breeds FIBROSIS
- 98. INFARCT
- 99. PRIMARY TUMORS (RARE) • HEMANGIOMA • LYMPHANGIOMA • fibroma • osteoma • chondroma • LYMPHOMA
- 100. MISC • Congenital Absence (very rare) • “Accessory” spleens (very very common, especially with splenomegaly!) •RUPTURE
- 101. THYMUS • Mother of all T-Cells • Massive in newborns, virtually absent in the elderly, bilobed • Under manubrium • 1) Thymocytes • 2) Epithelial Ret. Cells • 3) Hassal’s Corpuscles
- 102. HASSAL’s CORPUSCLES
- 103. DISEASES • HYPOPLASIA/APLASIA – DiGeorge Syndrome (i.e., velocardiofacial, 22q11.2 deletion) • CYSTS (incidental) • THYMOMAS
- 104. THYMOMAS • ALL (most) thymomas show counterparts of BOTH lymphoid as well as epithelial reticular cells, hence, the classic name “LYMPHOEPITHELIOMA” – Benign thymoma: (encapsulated) – Malignant Thymoma I: (locally invasive) – Malignant Thymoma II: (easily metastasizable)
- 105. THYMOMAS
Hinweis der Redaktion
- Classical features of peripheral white cells, recognition algorhythms
- Linear topics
- Grouped topics
- Classical features of peripheral white cells, recognition points.
- Many names, same cell
- ALL blasts look the same on routine stains, whether they are myeloblasts, lymphoblasts, monoblasts, etc. A blast is a blast is a blast is a blast is a blast is a blast is a blast is a blast is a blast is a blast is a blast is a blast is a blast! Please remember you have all been conditioned to think a NUCLEOLUS is darker than the rest of the NUCLEUS, as it is on H&E, but in the usual stains we stain bone marrow smears or peripheral smears with, i.e., Wright or Giemsa respectively, the nucleoli are LIGHTER!!!
- MYELOPEROXIDASE stains are often use to identify cells believed to be of MYELOID origin, such as blastic looking cells, because you cannot really tell for sure on Wright’s or Giemsa stains. Are these substances part on innate or learned immunity? ANS: INNATE
- Notice the parallel with anemia and thrombocytopenia. Here is the tip of the iceberg: acetazolamidealloprinol, asparaginasecaptopril, carbamazepine, cephalosporins, chloramphenicol, chlordiazepoxide, chlorpropamide, chlorthalidone, cimetidine, cyclophosphamide, ethacrynic acid, fluorouracil, furosemide, gold salts, ibuprofenimipramine, indomethasone, meprobamate, methimazole, methotrexate, metronidazole, nitrofurantoin, penicillamine, penicillins, phenothiazines, phenylbutazone, phenytoin, procainamide, procarbazine, propylthiouracil, quinidine, quinine, rifampin, spironolactone, sulfonamides, sulindac, thioridazine, tolbutamide,k Vancomycin
- TOXIC GRANULES are EXAGGERATIONS of the marrow’s normal granularity, DOHLE bodies are fragments of remaining dilated rough ER. Neutrophilia can be viewed as a NONSPECIFIC index of acute infection, especially bacterial, but also tissue necrosis.
- Following activation by an immune stimulus, eosinophils degranulate to release an array of cytotoxic granule cationic proteins that are capable of inducing tissue damage and dysfunction. These include: 1) major basic protein (MBP) 2) eosinophil cationic protein (ECP) 3) eosinophil peroxidase (EPO) 4) eosinophil-derived neurotoxin (EDN)
- Not only are basophils RARE to find normally, but pure “basophilia” is also VERY rare. When activated, basophils degranulate to release histamine, proteoglycans (e.g. heparin and chondroitin), and proteolytic enzymes (e.g. elastase and lysophospholipase). They also secrete lipid mediators like leukotrienes, and several cytokines
- Why would monocytosis be linked to granulomatous diseases? Answer: Monocytes are macrophages in circulation, and granulomatous diseaseas are macrophage diseases. Is it surprising that many classical granulomatous diseases are also characterized by monocytosis, because macrophages are the CHIEF cells of granulomas? Answer: NO, not surprizing, it would be logical even!
- Would it be a fair statement to say that whereas, neutrophilia is characterized by bacterial infections, lymphocytosis can be an index of many viral infections? Answer: Yes, it is fair, but there are many exceptions.
- EXTRAMEDULLARY HEMATOPOESIS is most common in the spleen, liver, and lymph nodes. Now we move from increases of peripheral blood leukocytes to increases of marrow in general, increases in cellularity (one dimension of expansion), increases from axial to appendicular skeleton (second dimension of expansion), and increases into other organs which do not normally make marrow in adults, such as liver and spleen and lymph nodes (third dimension of expansion).
- CML is the CLASSIC prototype of all myeloproliferative diseases.
- If a CML had much more than 10% blasts, you might suspect that the patient was going into a “blast crisis”.
- This marrow is virtually 100% cellularity!!! This is the HALLMARK of CML, and all the cells are still marrow cells although blasts are INCREASED, i.e., more than 1-2 %. The “spaces” are NOT fat, they are blood vessels. This marrow is virtually 100% cellularity!!! This is the HALLMARK of CML, and all the cells are still marrow cells although blasts are INCREASED, i.e., more than 1-2 %. The “spaces” are NOT fat, they are blood vessels.
- This marrow is virtually 100% cellularity!!! This is the HALLMARK of CML, and all the cells are still marrow cells although blasts are INCREASED, i.e., more than 1-2 %. In this CML megakaryocytes are proliferating so what OTHER myeloproliferative disease could this be confused with? Ans: essential thrombocythemia (also called essential thrombocytosis)
- This marrow is about 90% cellular and looks a lot like CML. This is P. vera
- Note that this is NOT a marrow biopsy but a smear, so TRUE marrow cellularity cannot be assessed, however MOST of the cells are megakaryocytes!
- Leukoerythroblastosis is an anemic condition resulting from space-occupying lesions in the bone marrow and characterized by the presence of immature granular leukocytes and nucleated erythrocytes in the circulating blood. Also called myelophthisic anemia.
- Note most of the marrow looks “fibrotic”. What stain could help you confirm that this is fibrous tissue? (trichrome would stain collagen green)
- CHRONIC leukemias, are essentially DIFFERENT from ACUTE leukemias, sorta.
- The most life saving thing you can learn today is how to recognize a blast! HUGE NUCLEUS NUCLEOLI (stain LIGHTER not DARKER than the rest of the nucleus on Wright stain), How many nucleoli does that one blast cell have? Answer: 3 NO cytoplasmic differentiation NOBODY IS GETTING OUT OF THIS ROOM ALIVE UNTIL THEY CAN IDENTIFY A BLAST CELL!
- Ataxia-Telangiectasia is characterised by: Early-onset progressive cerebellar ataxia (difficulty with control of movement) Ocular apraxia (difficulty following objects across visual field) Telangiectasias of the eyes and skin Immunodeficiency, low immunoglobulin concentrations Chromosomal instability Hyper-sensitivity to ionizing radiation Increased incidence of malignancies (primarily hematologic). Raised alpha-fetoprotein levels.[4] Absent thymic shadow on X-ray. Ovarian dysgenesis
- I will admit, these “blasts” DO look a little lymphocytic.
- You can usually diagnose CLL simply from a CBC printout, but should verify the cells visually.
- Many cells from CLL have a “smudge” or “basket” appearance. If you know what a NORMAL lymphocyte looks like, you can diagnose CLL purely by numbers! No marrow exam needed!
- Why would a CLL have hypogammaglobulinemia? ANS: LYMPHS are the precursors of plasma cells!
- An ironic thing is that the identification of plasma cells in the marrow for myeloma depends on how much they resemble NORMAL plasma cells! But do think the amount of monoclonal protein depends on how much they look and act like normal plasma cells? Ans: YES
- Please memorize those THREE diagnostic features of plasma cells, the malignant plasma cells of MM look like normal plasma cells usually. Ironically, most myelomas are recognized because they look like NORMAL plasma cells!
- Normal on left, myeloma on right. Batman sign?
- *The presence of large proliferations of monoclonal immune proteins are believed to interfere with normal coagulation.
- Note the “lytic” lesions. Often the term “punched out” is used also.
- What if you have a plasmacytoma but NO monoclonal gammopathy?
- What if you have a monoclonal gammopathy but NO demonstrable proliferation of plasma cells?
- What is the difference between a monoclonal and a polyclonal gammopathy? (monoclonals usually show a SPIKE on serum protein electrophoresis, SPE). Monoclonals are the result of malignant proliferations, polyclonals are generally the result of chronic inflammation.
- Well, after CML, CLL, and ALL, all that is left is the BIG ONE! Lets make this REAL easy…..ALL blasts look alike!!!! Why? Because there is NO cellular differentiation beyond the primitive stem cell appearance! Don’t confuse M (myeloid), with simply M, myelocytic series!
- AML’s are classified according to where the arrest occurs, i.e., the cells which proliferate in marrow and peripheral smear, and organs.
- Blasts, blasts with AUER rods. Auer rods are clumps of azurophilic granular material that form elongated needles seen in the cytoplasm of leukemic blasts. They are composed of fused lysosomes and contain peroxidase, lysosomal enzymes, and large crystalline inclusions.
- Acute promyelocytic leukemia, remember promyelocytes have BOTH nucleoli AND nonspecific granules, true BLASTS do NOT have granules. Do you remember that M3 has a high degree of association with DIC?
- In AMML, M4, many of the peripheral leukemic cells look like monocytes, while in M5, Acute Monocytic Leukemia, MOST of them look like monocytes. M5 has also been called “Schilling”-type leukemia. (NOT the same Schilling of the B12 Schilling test)
- In M6, many of the cells may resemble erythroid cells, in M7, many of the cells may resemble megakaryocytes. But in reality you would probably never think the blasts of M7 are related to megakaryocytes.
- Note AMAZING similarity to ALL, clinically. Surprised? Ans: NO CCPP
- Know the difference between a myelo-”proliferative” and a myelo-”dysplastic” disease.
- Why does the phrase “effaced architecture” appear on my pathology reports of malignant lymphomas?
- BENIGN FOLLICULAR HYPERPLASIA. Larger and more numerous than normal follicles. MEDULLA may be compromised.
- BENIGN SINUS HISTIOCYTOSIS. The cortical area may be compromised. SINUS HISTIOCYTOSIS may be seen in reaction to cancer, even if there are NO tumor cells in the lymph node.
- Mention Steve Swerdlow and link to his father!
- Why does the pathologist often use the word “buttocks” cell, to describe a “cleaved” cell lymphoma?
- “HAIRY” cell leukemia/lymphoma consists of lymphocytes which look hairy, but remember, on SEM ant TEM they all look hairy!
- “Effacement” means any pattern in which follicles in the cortex and cords/sinuses in the medulla cannot be recognized because they are “effaced” or “obliterated”
- Most pathologists HATE lymphoma classifications with a passion!
- I HATE this slide.
- I HATE this slide even worse.
- If you feel the need to memorize this fine, but better to remember: T markers <10 B markers 19-23 Mono/Mac teens RS 15, 30
- Prognosis of HD disease is related directly of percentage of lymphocytes and inversely to number of RS cells. Prognosis of HD disease is related directly of percentage of lymphocytes and inversely to number of RS cells. Prognosis of HD disease is related directly of percentage of lymphocytes and inversely to number of RS cells. Prognosis of HD disease is related directly of percentage of lymphocytes and inversely to number of RS cells. Prognosis of HD disease is related directly of percentage of lymphocytes and inversely to number of RS cells.
- STERNBERG REED cells are called “lacunar” cells in one of the most common forms of HD called NODULAR SCLEROSING
- If there was only one tiny microscopic focus of metastatic tumor cells in this lymph node, where would it most likely be?
- The subcapsular sinus of the lymph node is the FIRST place you will spot a metastatic tumor nest!
- We can also use the 50/50 principle generally for the spleen too: 50% “red” pulp, 50% “white” pulp. RED PULP = RBCs (grossly and microscopically) WHITE PULP = LYMPHS (grossly only, BLUE, microscopically)
- Notice the “confluence” of WHITE pulp? Could this be lymphoma involvement? Ans: Yes Could this be granulomas? Ans: YES
- Portal hypertension, prominence of RED pulp, i.e., red pulp >> 50%
- What 3 malignancies have the highest preference for splenic metastases? malignant melanoma lymphomas testicular germ cell tumors
- Why is a splenic infarct a “pale” infarct? Answer: single end-artery blood source
- Note all these are benign, except fot the lymphoma. The commonest MALIGNANT tumor primary to the spleen is a LYMPHOMA, by far!
- Are you more likely to see accessory spleens with splenomegaly? Ans: YES
- Hassal’s corpuscles are fused epithelial reticular cells
- Benign (encapsulated), Malignant I (invasive), and Malignant II (easily metastasizable) Note that the classification of thymomas has little to do with the appearance of the cells, but in the BEHAVIOR of the tumor grossly: 1) Encapsulated 2) Invasive 3) Metastatic