1. A Quantum Framework for Determining Rotational-Vibrational Spectra and Stability
of Diatomic Molecules
Jacovie Rodriguez
Department of Physics and Brain & Cognitive Sciences, 372 Memorial Drive, Cambridge, MA, 02139
(Dated: July 17, 2015)
The study of diatomic molecules is a step of complexity beyond the quantum Hydrogen atom
model, potentially offering a physical explanation for descriptions seen in chemical reactions. How-
ever, even the simplest diatomic case is rather complex, since there are interactions between the
electrons of one atom and the nuclei of the other atom. The degrees of freedom that separate
diatomic molecules from the single-atom potentials are the descriptions of rotation and vibration,
which are derived from considering the kinetic energy of the nuclei. Rotation can initially be ap-
proximated as if the nuclei were point masses rotating like a dumbbell, with correction terms coming
from centrifugal effects. Vibration can initially be approximated as a quantum harmonic oscillator,
with correction terms coming from anharmonicities in the potential. The constraints that arise from
the consideration of these energies give a bound on the stability of a diatomic molecule and its bond
length, with robust results for the simplest diatomic molecule, H2.
I. INTRODUCTION
Historically, chemistry has characterized molecular
interactions through experimentation, but quantum
physics is necessary for truly understanding the physi-
cal causes behind these interactions. For example, the
quantum model of the Hydrogen atom gave a full math-
ematical description of the electron orbitals, uniting a
physical and a chemical theory. The next order of com-
plexity that could result in a similar unification is analysis
of the set of diatomic molecules.
The single-atom system was straightforward to ana-
lyze because we treated the nucleus as stationary, and
the electron as evolving through time in a radially sym-
metric potential. Since radial potentials have eigenstates
that are also eigenstates of angular momentum, the so-
lution to this system is simply an analysis of this oper-
ator. However, analyzing the interactions of two atoms
becomes far more complex due to multiple interaction
terms.
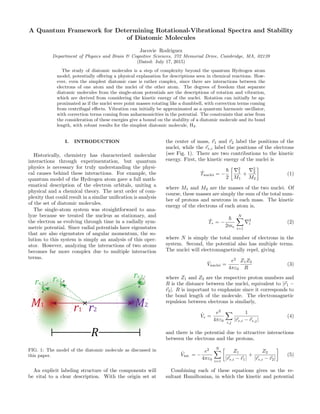
FIG. 1: The model of the diatomic molecule as discussed in
this paper.
An explicit labeling structure of the components will
be vital to a clear description. With the origin set at
the center of mass, r1 and r2 label the positions of the
nuclei, while the re,i label the positions of the electrons
(see Fig. 1). There are two contributions to the kinetic
energy. First, the kinetic energy of the nuclei is
ˆTnuclei = −
2
2
1
M1
+
2
2
M2
(1)
where M1 and M2 are the masses of the two nuclei. Of
course, these masses are simply the sum of the total num-
ber of protons and neutrons in each mass. The kinetic
energy of the electrons of each atom is,
ˆTe = −
2me
N
i=1
2
i (2)
where N is simply the total number of electrons in the
system. Second, the potential also has multiple terms.
The nuclei will electromagnetically repel, giving
ˆVnuclei =
e2
4πε0
Z1Z2
R
(3)
where Z1 and Z2 are the respective proton numbers and
R is the distance between the nuclei, equivalent to |r1 −
r2|. R is important to emphasize since it corresponds to
the bond length of the molecule. The electromagnetic
repulsion between electrons is similarly,
ˆVe =
e2
4πε0 i,j
1
|re,i − re,j|
(4)
and there is the potential due to attractive interactions
between the electrons and the protons,
ˆVint. = −
e2
4πε0
N
i=1
Z1
|re,i − r1|
+
Z2
|re,i − r2|
(5)
Combining each of these equations gives us the re-
sultant Hamiltonian, in which the kinetic and potential
2. 2
terms will be grouped.
ˆHdia. = ˆTnuclei + ˆTe + ˆVnuclei + ˆVe + ˆVint.
≡ ˆTdia. + ˆVdia. (6)
While this form of the Hamiltonian is descriptive of the
system, and also generalizeable to higher-number atomic
molecules, the multiple degrees of freedom can quickly
obscure the energy eigenstates. This paper will analyze
the diatomic molecule with the goal of simplifying the
degrees of freedom while still giving an accurate spec-
tra of the states. A successful framework of diatomic
molecules will lead to understanding a key property of
these molecules: stability.
II. METHODS OF SOLVING THE
SCHR¨ODINGER EQUATION
An appropriate approximation begins by constructing
quantities involving the electron that are small enough
to be negligible at high order. For example, an obvious
quantity is the ratio of electron mass to proton mass,
which is ∼ 5 × 10−4
. The electrons are quickly able
to adjust their position relative to nuclei adiabatically
since the nuclei are orders of magnitude more massive [2].
Thus, the analysis of this system will treat the molecule
as rigid (which will be comparable to the analysis of the
Hydrogen molecule), and treat the kinetic motion of the
nuclei as a perturbation.
ˆH0 = ˆTe + ˆVdia.
ˆδH = ˆTnuclei (7)
In this separation, the eigenstates of ˆH0 assume that the
nuclei maintain a constant distance, while the eigenstates
of ˆδH let the positions of the nuclei change. This will
allow us to create separable solutions of the wavefunction,
which will act as a function of the nuclei positions ri and
the electron positions re,i.
ψdia.(ri, re,i) = ξ(re,i; R)χ(ri) (8)
Here, ξ refers to the states dealing with the electric inter-
actions and kinetic motion of the electrons. Naturally, it
takes the positions of each particle as a parameter, but
note that the positions of the nuclei in this wavefunction
are fixed, meaning that R = |r1 −r2| is a fixed parameter
of this wavefunction. The motion of the nuclei is instead
reserved for the wavefunction χ, which directly takes the
nuclei positions as its parameters. The coordinate sys-
tem as defined in the outset (Fig. 1) takes these positions
relative to the center of mass, so there are two possible
types of motion that describe the nuclei motion: rotation
and vibration. Thus, the kinetic energy of the nuclei will
be defined by these degrees of freedom as
Tnuclei = Erot. + Evib. (9)
The perturbation will not effect the ξ states, so the action
of the Hamiltonian on ξ simply comes from ˆH0, which
involves the kinetic energy of the electrons and the po-
tential from the positions of the charged particles. Note
that the number of electrons is an important parameter
in both of these energy contributions, so N – the total
number of electrons – will be a state that determines the
energy. Therefore, the first-order energy from ˆH0 on ξ is,
ˆH0ξn = E(0)
n ξn = ( ˆTe + ˆVdia.)ξn
=⇒ E(0)
n = ˆTe + Vdia(R)
(10)
The important result of Eq. 10 is that the only mean-
ingful position parameter affecting the energy in the ξ
states is the distance between the nuclei, which essen-
tially means that the general orientation of the molecule
does not affect its rigid description. The orientation-
dependent physics comes from looking at the χ states.
Rotation corresponds to changes in θ and φ, while vibra-
tion corresponds to changes in r. Since the kinetic energy
of the nuclei is separated into these states by Eq. 9, the
wavefunction is separable into
χ(r, θ, φ) = β(r)ρ(θ, φ) (11)
where ρ represents the rotational states and β represents
the vibrational states.
A. Solving for Rotations
1. Rigid Rotor Model
The first step towards describing the nuclei rotations
is the “rigid rotor” model, where the primary motion is
just rotational. We expect these modes to have a bigger
effect than vibrational states, since rotations comprise
more dramatic motions than vibrations which are smaller
than R. The model will give solutions to the ρ(θ, φ).
The rigid rotor model takes the nuclei and approx-
imates them as point masses attached by a weightless
rod. This approximation resembles the classical problem
of a dumbbell with two weights on the end, and just as
the classical example uses the concepts of center of mass
and moment of inertia, so will this quantum model.
Placing the origin of our coordinates at the center of
mass will simplify the calculations. The center of mass
is described by RCM = (M1R1 + M2R2)/(M1 + M2), so
the distances to the nuclei can be described by ratios of
the total bond length R.
R1 =
M2
M1 + M2
R
R2 =
M1
M1 + M2
R (12)
The moment of inertia is easy to write with the point
3. 3
FIG. 2: The first step towards understanding χ states is the
rigid rotor model, which only has rotations about its center
of mass.
masses,
I =
2
i=1
MiR2
i =
M1M2
M1 + M2
R2
= µR2
(13)
where µ is the reduced mass.
The moment of inertia has a clear connection to two
operators already commonly used. First, for a given ro-
tation speed ω, the kinetic energy operator becomes,
ˆTrot. =
Iω2
2
(14)
Second, the definition of angular momentum L = Iω
combined with (14) gives the definition,
ˆL2
rot. = 2I ˆTrot. = 2µR2 ˆTrot. (15)
This simplifies the Hamiltonian dramatically. The only
energy here is the kinetic, so ˆHrot. = ˆTrot., and Eq. (15)
means the eigenstates of the Hamiltonian are just eigen-
states of the angular momentum squared; i.e.,
ρ(θ, φ) = 2µR2
Ym, (θ, φ) (16)
These eigenstates are the spherical harmonics, which are
described by L2
= ( + 1) 2
. The energy eigenvalues of
rotation are:
ˆL2
rot. = 2µR2 ˆTrot.,
=⇒ ( + 1) 2
= 2µR2
Erot.,
Erot., =
( + 1) 2
2µR2
(17)
One useful application of these rotational states is that
often, when a diatomic molecule is radiated with photons,
the energy of the photon will be absorbed into a rota-
tional state. These transitions between states are linear
in , as we see:
∆Erot., = Erot., +1 − Erot., =
( + 1) 2
µR2
(18)
The energy transitions are diagrammed in Fig. 3, which
demonstrates that the energy spectrum seen from emis-
sions should be peaked with an equal space of ∆Erot.,0
between each peak. One useful application of this is being
able to measure the bond length of a diatomic molecule
from its rotational spectrum. If the difference between
peaks is measured to be an experimental value ˜E, then
the bond length of that diatomic molecule is given by,
R =
2
µ ˜E
(19)
The assumption of rigidity is sufficient for slower ro-
tations, but as the energy levels increase, the rigidity of
the molecule cannot hold and thus R will increase.
FIG. 3: The energy bands of the first few rotational states
in the rigid motor model, scaled to units of 2
/2µR2
. The
transitions between bands are linear in and can be used to
determine the bond length of diatomic the molecule.
2. Centrifugal Correction
In the outset of the rigid rotor model, the adiabatic
condition held that the electrons would maintain their
4. 4
relative position to the nuclei throughout its motion.
While this is a good approximation, it breaks down when
the atom has a high rotational energy because with rapid
rotation, the electrons will centrifugally spread radially
away from rotation. The classical picture would posit
this situation as a force due to radial acceleration, with
magnitude
Fr = µω2
R =
( + 1) 2
µR3
(20)
where the angular velocity ω is connected to the kinetic
energy and thus angular momentum eigenvalues. The
counterbalancing force would be due to the potential be-
tween the nuclei, going as
Fb = −∂ΓVnuclei(Γ) (21)
where Γ represents the new stretched distance between
the nuclei.
Note that in general, Γ will be small, so expanding by
a Taylor Series around the set distance R results in,
Vnuclei(Γ) =
∞
k=0
1
k!
∂k
Γ [Vnuclei(Γ)]|Γ=R (Γ − R)
k
(22)
The potential must be at a local minimum around
R since this is essentially the equilibrium-point dis-
tance of the nuclei at the zero-rotation point. Thus,
∂1
Γ [Vnuclei(Γ)]|Γ=R must be zero and the first-order term
can be neglected. The approximation will take this po-
tential energy to the quadratic term, then neglect the
higher orders. Importantly then, the general result is
Vdia.(r) = Vdia.(R) +
1
2
∂2
r [Vdia.(r)]|R (r − R)
2
(23)
With the potential as given by Eq. 23, the force as de-
fined in Eq. 21 must be linear up to a balancing constant
b,
Fb = −b(Γ − R) ˆR (24)
The magnitudes of the two forces must balance, equating
Eq. 24 and Eq. 20.
( + 1) 2
µR3
= b(Γ − R)
Γ = R +
( + 1) 2
bµR3
(25)
The result is promising, since Γ = R when there is no
rotation, and Γ increases when there is rotation. Thus,
this contribution to the energy is
Erot. =
( + 1) 2
2µR2
+
1
2
b(Γ − R) (26)
More precisely, the energy contributions can be expanded
since Γ is small. The value x = ( +1) 2
bµR3 1, so Γ can
be rewritten as
Γ = R(1 + x)
=⇒
1
Γ2
=
1
R2
1
(1 + x)2
=
1
R2
1 − 2x + 3x2
+ O(x3
)
=⇒ Γ ≈
R
√
1 − 2x + 3x2
(27)
Finally then, the contribution to the energy is
Erot. =
( + 1) 2
2µR2
+
Rb
2
1
1 − 2 ( +1) 2
bµR3 + 3 2( +1)2 4
b2µ2R6
− 1
(28)
The form of the stretching contribution in Eq. (27)
has a local maximum of
Γmax = R 3/2 (29)
at x = 1/3. Even at fast rotational speeds, the bond
length only increases about 22%. The corresponding shift
in energy is an increase by Rb
2 3/2 − 1 , so the en-
ergy correction at maximum bond length increases lin-
early with the balancing constant.
B. Solving for Vibrations
1. Radial Equation
The χ states in Eq. 11 are separated by the vibra-
tional and rotational states. A strong connection has
been made between the form of these solutions and the re-
sults obtained from the radial potentials in simpler cases.
Having already discussed the rotational states, we now
focus on the vibrational states β(r).
The action of the Hamiltonian on these states is of the
same form,
ˆHβ(r) = E0
n + ˆTnuclei β(r)
= −
2
2µ
2
+ Vdia.(R) β(r)
(30)
Since β states strictly depend on the radial coordinates,
only the first term of the radial form of 2
, with deriva-
tives in r, will be kept.
2
r =
1
r2
∂
∂r
r2 ∂
∂r
(31)
The others refer to rotational states, already discussed.
To reduce ambiguity, r is the variable used to indicate
that this variable changes, but in fact refers to the same
5. 5
quantity of internuclear distance. Thus, the Schr¨odinger
Equation takes on the form of the radial equation,
1
r2
d
dr
r2 dβ(r)
dr
+
2µ
2
Evib. − Vdia.(r) −
( + 1) 2
2µr2
β(r) = 0
(32)
Now, quite a few approximations will be used to ana-
lyze this equation. In the simplest case, vibrations occur
without any rotational motion, meaning = 0. Eq. (23)
revealed that the potential is, in general, well approxi-
mated by a quadratic form in R when the vibrations are
small in comparison to the rest internuclear distance. A
quadratic potential is the form of the quantum harmonic
oscillator (QHO), which will be the first approximation
of the vibrational states.
The energy eigenstates of the harmonic oscillator are
characterized by number states of Ek = ω k + 1
2 where
k is in the set of nonnegative integers (k = 0, 1, 2, . . .)
[8]. Remember that in this case, r parametrizes a 1D
oscillation. Straightforwardly then, the vibrational states
can be written as
βk(r) =
1
√
2kk!
µω
π
1/4
e−µωr2
/2 2
Hk
µω
r (33)
where the Hk are the Hermite polynomials as defined by
Hk(x) = (−1)k
ex2 dk
dxk
e−x2
(34)
and the energy eigenvalues of vibration are simply
Evib.,k = ω k +
1
2
(35)
The form of these states and the corresponding energy
values each contain the unknown parameter ω. This pa-
rameter is not arbitrary, but in fact specifically depends
on the potential similar to the standard QHO,
ω =
b
µ
=
∂2
r [Vdia.(r)]|R
µ
(36)
2. Large Vibration Corrections – The Morse Potential
The assumption that the potential is essentially
quadratic around the rest internuclear distance only
holds provided that the stretching length is not large.
However, this is not always going to be the case, espe-
cially if a high amount energy goes into vibrations. The
quadratic approximation to the potential is a poor de-
scription when far from R, so instead the analysis must
turn to a better approximation. This will rely on the
Morse Potential,
Vdia.(r) ≈ VMorse(r) = E∞ 1 − e−γ(r−R)
2
. (37)
The Morse potential is an improved approximation for
the diatomic molecule since it accounts for multiple im-
portant properties. The energy of vibrational states is
limited in the diatomic molecule because with excessive
vibration, the atoms would be too far separated to main-
tain the bond and the bond would break. The Morse
potential allows for these unbound states by approach-
ing a maximum energy E∞ as r → ∞. It is also ap-
parent that the bond length cannot ever be zero, since
the electromagnetic interactions between electrons of the
different atoms prevents the two atoms from contacting.
The Morse potential characterizes this by diverging to
infinity at r = 0 (see Fig. 4). The parameter γ here
r
V
Morse
V
VQHO
E∞
R
FIG. 4: The Morse Potential compared to the quadratic ap-
proximation of the Quantum Harmonic Oscillator. The Morse
Potential gives two important properties that the Quantum
Harmonic Oscillator does not. First, as r goes to infinity, the
potential plateaus to a constant value E∞. This allows un-
bound states with energy above E∞. Second, the potential
diverges to infinity as r goes to zero, which accurately repre-
sents the repulsion of the electrons preventing the atoms from
contacting.
characterizes the rate at which VMorse approaches E∞.
Connecting this to the quadratic approximation means
setting the parameter with the same “spring constant”
as Eq. (36), in that,
γ =
b
2E∞
=
∂2
r [Vdia.(r)]|R
2E∞
(38)
The Schr¨odinger Equation for this potential is,
−
2
2µ
∂2
∂r2
+ E∞ 1 − e−γ(r−R)
2
β(r) = Evib.β(r)
(39)
which has exact solutions derived in a similar operator
method to the harmonic oscillator [5]. From this poten-
tial, then, the energy contribution becomes,
Evib.,k = ω k +
1
2
−
2
ω2
4E∞
k +
1
2
2
(40)
6. 6
where the ω is still analogous to angular frequency of the
vibration, but now takes the form,
ω =
b
µ
= γ
2E∞
µ
(41)
Evib.
E = 2ћω∞ E = 3ћω∞ E = 4ћω∞ E = 5ћω∞
FIG. 5: The energy bands of the first few vibrational states
in the Morse Potential Model, in units of ω. The spectrum
is given for different values of E∞, demonstrating how the
potential determines a maximum vibrational energy, and the
increasing bands approach the maximum value quadratically.
The resulting form of the energy is quadratic in k,
meaning that there is a maximum energy that can be
stored in vibrational modes, supporting the notion that
the vibrational energies cannot go above a certain value
since the atoms would break the internuclear bond. The
maximum energy is found by setting the derivative of Eq.
(40) to zero, and gives the expected result
Evib.,max = E∞ (42)
as the potential was designed to do. The quantum num-
ber k takes on the value,
kmax =
2E∞
ω
−
1
2
(43)
Importantly, this analysis demonstrates that the poten-
tial gives a strict bound on the maximum energy that
can go into vibrational modes.
C. Simultaneous Rotation and Vibration
Now that rotation and vibration have been fully ana-
lyzed, the descriptions need to be combined. Between the
two, rotations will have greater effects on the energy dis-
tribution since the intermolecular distance cannot change
drastically but the molecule can spin arbitrarily fast. In
the simplest case, an adiabatic approximation dictates
that the speed is low enough to make the change in ki-
netic energy of the electrons ˆTe be infinitesimally small.
Similarly, the adiabatic approximation states that cen-
trifugal effects will be negligible, and with small vibra-
tions, the potential ˆVdia. will only experience infinitesimal
changes. Thus, the only meaningful changes that can oc-
cur are transitions between the rotational and the vibra-
tional states. This is encapsulated in the conservation
law:
∂
∂t
ˆTnuclei = 0
=⇒
∂
∂t
Erot. = −
∂
∂t
Evib.
(44)
With the approximation that rotational energies are
much larger than vibrational energies, the energy terms
can sum though the vibrational term was derived by let-
ting = 0 in Eq. (32). In the simplest of cases, where
energies are near the ground state, the diatomic molecule
acts as a spherical harmonic with radial vibration. Thus,
the state is a function of the quantum numbers , k so
that:
ˆTnucleiχ ,k = E ,kχ ,k (45)
E ,k =
( + 1) 2
2µR2
+
b
µ
k +
1
2
(46)
where b can be thought of as an internuclear restoring
constant, determined by
b = ∂2
r [Vdia.(r)]|R (47)
At higher energies, larger corrections will come into play
- the first of which are the centrifugal corrections to ro-
tation and the anharmonic corrections to the vibrations.
Thus, the higher order corrections result in energy states
with,
E ,k =
( + 1) 2
2µR2
+
bR
2
1
1 − 2 ( +1) 2
bµR3 + 3 2( +1)2 4
b2µ2R6
− 1
+
b
µ
k +
1
2
−
b 2
4µE∞
k +
1
2
2
(48)
The results of the previous discussions show that this
atom is bound to have both rotational transitions as de-
scribed by Eq. (18) and also a maximum vibrational en-
ergy as given by Eq. (42). Disastrous for the atom, then,
7. 7
is if the rotational energy decreases by an energy level,
only to conserve this energy by raising the vibrational en-
ergy beyond its limit and breaking the internuclear bond.
This can occur if,
E∞ ≤
2
µR2
(49)
The value of E∞ depends on the configuration of
the charged particles in the diatomic molecule, while µ
depends on their masses. Take the simplest diatomic
molecule, H2 (dihydrogen), as an example. Suppose both
Hydrogen atoms have electrons in the ground state, then
E∞ (setting the potential to be zero at the intermolecular
distance) is roughly
E∞,H2 = Vdia. =
e2
4π 0
2
R
−
2
a0
(50)
where a0 is the Bohr’s radius. Reversing the inequality
from Eq. (49), the intermolecular distance of a stable di-
hydrogen molecule would have to approximately be larger
than
RH2
a0
2
1 + 1 −
4π 0
2
a0e2µ
(51)
These variables take on the following values in the case
of the dihydogren molecule: a0 = 52.9 pm, 0 = 8.854 ×
10−24
F/pm, = 1.055 × 10−34
J s, e = 1.602 × 10−19
C, and µ = 1.67 × 10−27
kg. Eq. (51) with these values
gives a theoretical value of
RH2
52.9 pm (52)
In fact, the experimental value of the dihydrogen
molecule bond length is 74 pm, so the approximation is
in fact rather close [7]. Using the centrifugal correction
in Eq. (29) brings the theoretical value to,
ΓH2,max =
3
2
RH2
64.8 pm (53)
which is even closer to the experimental value.
III. CONCLUSION
The diatomic molecule potential contains several fea-
tures not seen in the single-atom case. Electron motion
is minimal in contributing to energy due to the electron
mass being negligibly smaller than the proton mass, so
the significant contributions to the energy come from
changes in the motion of the nuclei. This kinetic energy
is characterized by two degrees of freedom: rotation and
vibration. The rotational states show a linearly increas-
ing energy transition, giving a way to measure the bond
length of the atom. Fast rotations also cause a centrifu-
gal pushing effect, slightly increasing the intermolecular
bond distance. Considering the quadratic nature of the
potential around the rest distance gives an approxima-
tion to the maximum stretching.
The vibrational states show a quadratically decreasing
set of energy levels that are constrained by the potential
of the atom. Together, the conservation of kinetic energy
shows that transitions between rotational and vibrational
energies place a lower bound on the intermolecular bond
distance. Calculating this bound for the simplest case,
H2, shows that this quantum mechanical framework sup-
ported experimental data.
In summary, systems with added degrees of freedom
often have more constraints due to the complexity of in-
teractions. Approximations on these interactions, even
while rudimentary, still give robust support to a theoret-
ical framework of understanding why diatomic molecules
have properties such as bond lengths and stability.
[1] Basdevant, J. L., and J. Dalibard. “Unstable Diatomic
Molecule.” The Quantum Mechanics Solver: How to Ap-
ply Quantum Theory to Modern Physics. Berlin: Springer,
2006. 11-16. Print.
[2] “CODATA Value: proton-electron mass ratio.” The NIST
Reference on Constants, Units, and Uncertainty. US Na-
tional Institute on Standards and Technology. 2011.
[3] DeKock, Roger L., and Harry B. Gray. “Net Bonding in
Molecules with 1s Valence Atomic Orbitals.” Chemical
Structure and Bonding. Menlo Park, CA: Benjamin/Cum-
mings Pub., 1980. 199. Print.
[4] Demtr¨oder, Wolfgang. “Diatomic Molecules.” Atoms,
Molecules and Photons: An Introduction to Atomic-,
Molecular-, and Quantum-Physics. Berlin: Springer, 2006.
321-71. Print.
[5] Ta¸seli, H. “Exact Solutions for Vibrational Levels of the
Morse Potential.” Journal of Physics A: Mathematical and
General. 31.2 (1998): 779-88. Web.
[6] McQuarrie, Donald A. Quantum Chemistry. Mill Valley,
CA: U Science, 1983. Print.
[7] Weast, Robert C., Melvin J. Astle, and William H. Beyer.
CRC Handbook of Chemistry and Physics: A Ready-
reference Book of Chemical and Physical Data. Boca Ra-
ton, FL: CRC, 1984. Print.
[8] Commonly, n is used to characterize these number states,
but this has been previously used to represent the total
number of electrons in this paper.
![2
terms will be grouped.
ˆHdia. = ˆTnuclei + ˆTe + ˆVnuclei + ˆVe + ˆVint.
≡ ˆTdia. + ˆVdia. (6)
While this form of the Hamiltonian is descriptive of the
system, and also generalizeable to higher-number atomic
molecules, the multiple degrees of freedom can quickly
obscure the energy eigenstates. This paper will analyze
the diatomic molecule with the goal of simplifying the
degrees of freedom while still giving an accurate spec-
tra of the states. A successful framework of diatomic
molecules will lead to understanding a key property of
these molecules: stability.
II. METHODS OF SOLVING THE
SCHR¨ODINGER EQUATION
An appropriate approximation begins by constructing
quantities involving the electron that are small enough
to be negligible at high order. For example, an obvious
quantity is the ratio of electron mass to proton mass,
which is ∼ 5 × 10−4
. The electrons are quickly able
to adjust their position relative to nuclei adiabatically
since the nuclei are orders of magnitude more massive [2].
Thus, the analysis of this system will treat the molecule
as rigid (which will be comparable to the analysis of the
Hydrogen molecule), and treat the kinetic motion of the
nuclei as a perturbation.
ˆH0 = ˆTe + ˆVdia.
ˆδH = ˆTnuclei (7)
In this separation, the eigenstates of ˆH0 assume that the
nuclei maintain a constant distance, while the eigenstates
of ˆδH let the positions of the nuclei change. This will
allow us to create separable solutions of the wavefunction,
which will act as a function of the nuclei positions ri and
the electron positions re,i.
ψdia.(ri, re,i) = ξ(re,i; R)χ(ri) (8)
Here, ξ refers to the states dealing with the electric inter-
actions and kinetic motion of the electrons. Naturally, it
takes the positions of each particle as a parameter, but
note that the positions of the nuclei in this wavefunction
are fixed, meaning that R = |r1 −r2| is a fixed parameter
of this wavefunction. The motion of the nuclei is instead
reserved for the wavefunction χ, which directly takes the
nuclei positions as its parameters. The coordinate sys-
tem as defined in the outset (Fig. 1) takes these positions
relative to the center of mass, so there are two possible
types of motion that describe the nuclei motion: rotation
and vibration. Thus, the kinetic energy of the nuclei will
be defined by these degrees of freedom as
Tnuclei = Erot. + Evib. (9)
The perturbation will not effect the ξ states, so the action
of the Hamiltonian on ξ simply comes from ˆH0, which
involves the kinetic energy of the electrons and the po-
tential from the positions of the charged particles. Note
that the number of electrons is an important parameter
in both of these energy contributions, so N – the total
number of electrons – will be a state that determines the
energy. Therefore, the first-order energy from ˆH0 on ξ is,
ˆH0ξn = E(0)
n ξn = ( ˆTe + ˆVdia.)ξn
=⇒ E(0)
n = ˆTe + Vdia(R)
(10)
The important result of Eq. 10 is that the only mean-
ingful position parameter affecting the energy in the ξ
states is the distance between the nuclei, which essen-
tially means that the general orientation of the molecule
does not affect its rigid description. The orientation-
dependent physics comes from looking at the χ states.
Rotation corresponds to changes in θ and φ, while vibra-
tion corresponds to changes in r. Since the kinetic energy
of the nuclei is separated into these states by Eq. 9, the
wavefunction is separable into
χ(r, θ, φ) = β(r)ρ(θ, φ) (11)
where ρ represents the rotational states and β represents
the vibrational states.
A. Solving for Rotations
1. Rigid Rotor Model
The first step towards describing the nuclei rotations
is the “rigid rotor” model, where the primary motion is
just rotational. We expect these modes to have a bigger
effect than vibrational states, since rotations comprise
more dramatic motions than vibrations which are smaller
than R. The model will give solutions to the ρ(θ, φ).
The rigid rotor model takes the nuclei and approx-
imates them as point masses attached by a weightless
rod. This approximation resembles the classical problem
of a dumbbell with two weights on the end, and just as
the classical example uses the concepts of center of mass
and moment of inertia, so will this quantum model.
Placing the origin of our coordinates at the center of
mass will simplify the calculations. The center of mass
is described by RCM = (M1R1 + M2R2)/(M1 + M2), so
the distances to the nuclei can be described by ratios of
the total bond length R.
R1 =
M2
M1 + M2
R
R2 =
M1
M1 + M2
R (12)
The moment of inertia is easy to write with the point](data:image/gif;base64,R0lGODlhAQABAIAAAAAAAP///yH5BAEAAAAALAAAAAABAAEAAAIBRAA7)